Phenylketonuria is one of the “aminoacidopathies”. An aminoacidopathy is a fancy medical term for a malfunction in the bodies’ ability to handle specific amino acids, in this case, phenylalanine.
But before we get too crazy, first things first… What is an amino acid? Amino acids are the building blocks of proteins. Each amino acid consists of a carbon atom that has four “things” hanging off of it: an amine group, a carboxylic acid group, a hydrogen atom, and finally an “R” group. The “R” group varies depending on the specific amino acid being discussed. For example, the “R” in phenylalanine is a benzyl group.
Amino acids link up together via “peptide bonds”. A peptide bond is simply the carboxylic acid group of one amino acid “hooking” up to the amine group of another amino acid. This linking occurs over and over again resulting in a protein that can be thousands of amino acids in length.
With that background, let’s discuss one specific amino acid, phenylalanine, and what happens when the body has difficulty using it – a condition called phenylketonuria.
An enzyme in the liver called phenylalanine hydroxylase is necessary to transform phenylalanine into another amino acid called tyrosine. In certain individuals, the genes on chromosome 12 that encode the hydroxylase enzyme are deficient. Mutations in both of your hydroxylase genes (ie: the one you got from mom and the one you got from dad) must be deficient for phenylketonuria to develop; in other words, one adequate gene can produce enough normal hydroxylase to handle a normal dietary phenylalanine load. However, when both genes are deficient, phenylalanine can accumulate to abnormally high levels in the blood.
It is important to note that phenylketonuria can occur even if the hydroxylase enzyme is normal! How is this possible? The answer is because the hydroxylase enzyme requires another molecule called tetrahydrobiopterin (try saying that 3 times fast!) to function properly. When the enzyme (dihydrobiopterin reductase) that produces tetrahydrobiopterin is deficient, it gunks up the entire system and slows the ability of phenylalanine hydroxylase to convert phenylalanine to tyrosine.
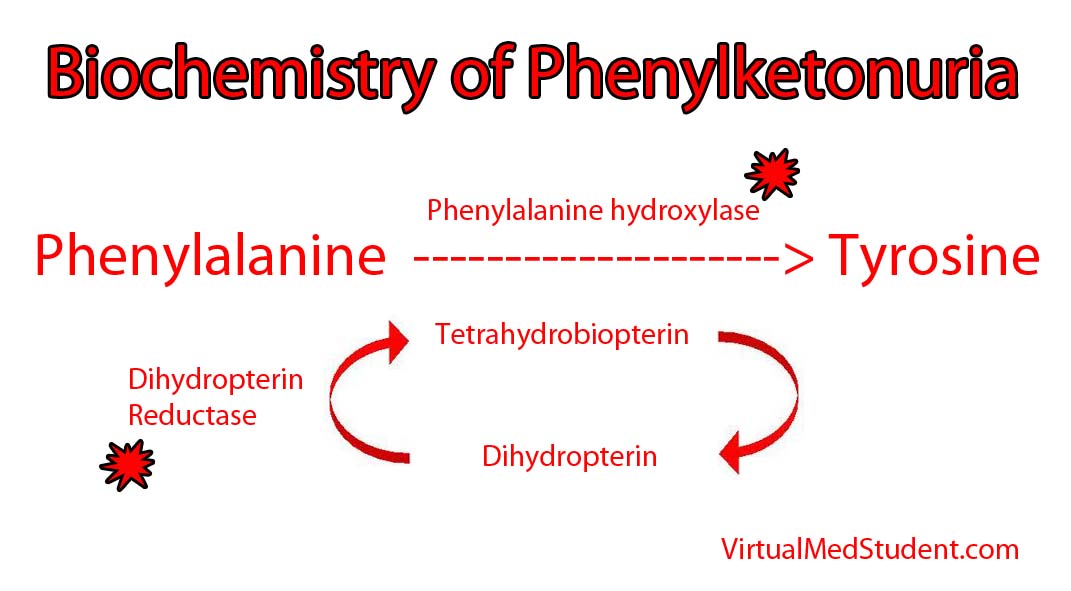
Either way you slice it, the excess phenylalanine interferes with the ability of other amino acids to enter the brain. This hinders brain development and, if left untreated, can cause mental retardation.
Signs and Symptoms
When excess phenylalanine backs up in the blood stream it blocks amino acid transporters in the brain. As a result, the brain does not get access to other essential amino acids, and patients that are left untreated, develop mental retardation and seizures.
Children affected by the condition also commonly have a musty odor. The odor is caused by phenylacetate, which is a breakdown product of excess phenylalanine.
Interestingly, patient’s who suffer from phenylketonuria are also usually blond haired and blue eyed. The reason for this is that tyrosine is necessary for melanin formation. Melanin is the pigment that gives hair, skin, and eyes their color. In phenylketonuria, phenylalanine is not converted to tyrosine and therefore melanin is not produced. The resultant lack of pigment causes blond hair and blue eyes.
Diagnosis
Many countries screen new babies for phenylketonuria because mental retardation can be prevented with strict dietary control. Diagnosis is based on finding an elevated phenylalanine to tyrosine ratio in the blood. Additionally, phenylpyruvate (a breakdown product of phenylalanine) will be elevated in urine samples of affected individuals.
Treatment
Treatment is, you guessed it, avoiding phenylalanine! This is done by ingesting a low protein diet, and in babies, using formula that is free of phenylalanine.
In phenylketonuria, tyrosine becomes an essential amino acid. Therefore, an important aspect of treatment is ensuring adequate intake of tyrosine via supplementation.
Overview
Phenylketonuria results when the body is unable to convert the amino acid phenylalanine into tyrosine. It can be caused by a primary genetic deficiency in the enzyme phenylalanine hydroxylase. It can also be caused by defects in the production of the hydroxylases’ necessary cofactor, tetrahydrobiopterin. Symptoms, if left untreated, include mental retardation. Patients with phenylketonuria are generally fair skinned and blue eyed because they have difficulty producing the melanin. Treatment is avoidance of phenylalanine and supplementation with tyrosine.
Related Articles
References and Resources
- Cunningham GC. Phenylketonuria – early detection, diagnosis and treatment. Calif Med. 1966 July; 105(1): 1–7.
- van Spronsen FJ, Huijbregts SC, Bosch AM, et al. Cognitive, neurophysiological, neurological and psychosocial outcomes in early-treated PKU-patients: A start toward standardized outcome measurement across development. Mol Genet Metab. 2011;104 Suppl:S45-51. Epub 2011 Oct 6.
- Mitchell JJ, Trakadis YJ, Scriver CR. Phenylalanine hydroxylase deficiency. Genet Med. 2011 Aug;13(8):697-707.
- Werner ER, Blau N, Thöny B. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem J. 2011 Sep 15;438(3):397-414. Review.