Insulin is a protein secreted by specialized cells in the pancreas known as β-cells. When the body senses a high level of glucose floating around in the blood it causes the pancreas to secrete insulin. Many individual insulin molecules then travel throughout the body where it binds to receptors on many different tissue types. For example, if insulin binds to hepatocytes (liver cells) it instructs those cells to remove glucose from the blood and store it in a polymerized form known as glycogen. Insulin’s goal is to bring blood sugar levels to within normal limits.
The problem in diabetes mellitus is that insulin is either not secreted by the pancreas, or it does not perform its functions appropriately. Either way, glucose is not removed from the blood stream. The result is a higher than normal blood sugar concentration.
There are two distinct forms of diabetes mellitus: type 1 diabetes (sometimes referred to as juvenile onset or insulin dependent diabetes) and type II diabetes (sometimes referred to as adult onset or insulin independent diabetes).
Type 1 diabetes is an auto-immune disorder, which means that the bodies’ immune system attacks itself. In this case, the body attacks the β-cells in the pancreas. Once enough β-cells are destroyed the pancreas can no longer secrete insulin.
The pathology of type 2 diabetes mellitus is more complicated. There is no single "cause" of type 2 diabetes, and as such, it is believed to be the result of a combination of factors. Genetics, lifestyle, and diet all play an important role in the development of this form of the disease. I like to think of type 2 diabetes as a slew of factors that have beaten down the bodies ability to properly manage blood sugar levels.
In type 2 diabetes the pancreas is still able to produce insulin. However, the problem is that insulin does not cause its normal effect on body tissues. This is known as "insulin resistance." In an effort to combat this resistance the pancreas secretes more insulin. Once the insulin resistance becomes too severe the pancreas can not keep up, and blood sugar levels begin to rise. If the levels rise beyond a specific laboratory threshold the diagnosis of diabetes is made.
How Does Diabetes Mellitus Present?
High blood sugar levels lead to the symptoms of diabetes mellitus. One of the most common symptoms of diabetes is a frequent need to urinate. The reason this occurs is because the excess blood sugar exceeds the kidney’s ability to re-absorb it. As a result, glucose leaks into the urine. The body is forced to dilute this excess solute load by secreting more water into the urine. Hence more urine production –> increased trips to the bathroom!
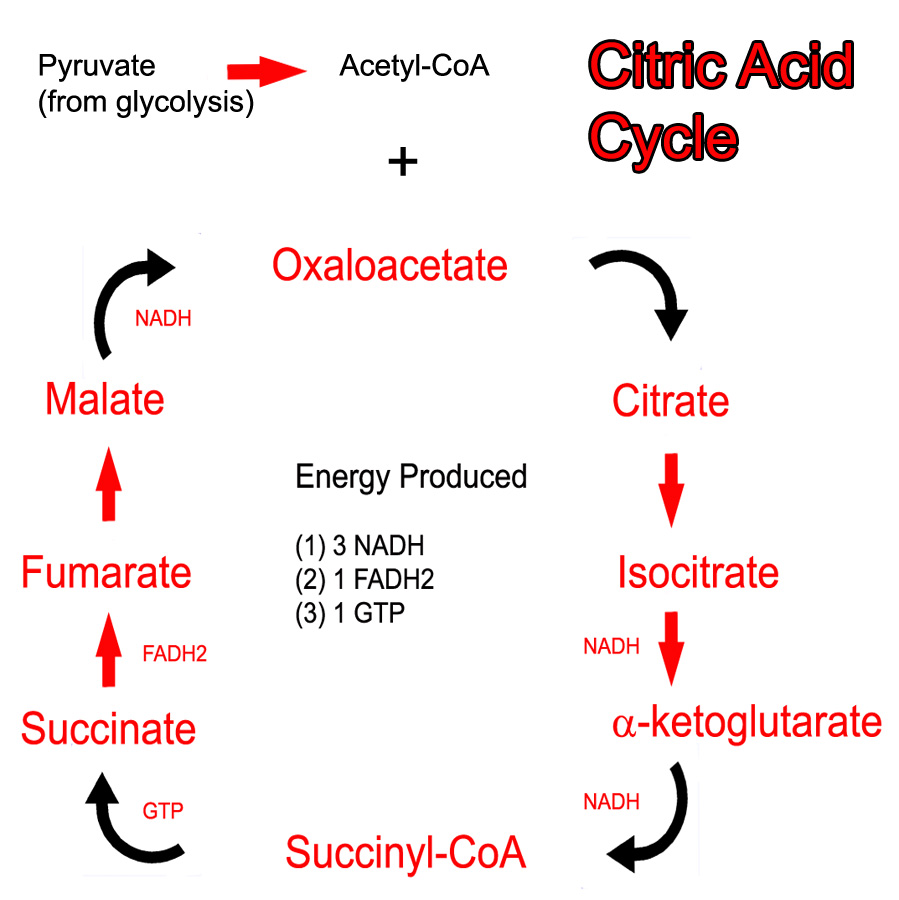
Since diabetics have difficulty using sugar as a fuel they often lose weight (although most type II diabetics are overweight or obese secondary to excessive caloric intake). Diabetes, in metabolic terms, is similar to slowly starving. The body is unable to utilize sugar appropriately, which is the main “fuel” for most people. Normally, excess sugar gets converted to fat (ie: for you “need-to-know it all types” glucose is converted into acetyl-CoA fragments. If these are not “burned” by the Krebs cycle they get polymerized into long chain fatty acids, which get stored in fat tissue). However, in diabetics the sugar is "lost" in the urine and not used for metabolic purposes.
is similar to slowly starving.
When a diabetic crisis occurs in type 1 diabetics it is referred to as "diabetic ketoacidosis". It is known as "non-ketotic hyperosmolar syndrome" if it occurs in a patient with type 2 diabetes.
What Else Can Happen to People with Diabetes?
There are many complications of diabetes mellitus. These complications are the result of years of high sugar levels in the blood. Over time the excess sugar undergoes a metabolic transformation known as "glycosylation". Glycosylated sugar is toxic to nerves and blood vessels.
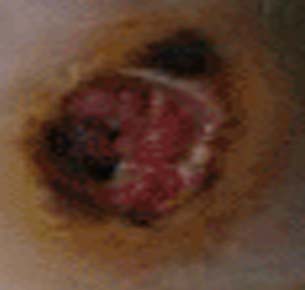
Diabetic foot ulcer
In addition, nerves that carry sensory information can also be damaged, especially at the finger tips and toes. Patients with this type of nerve damage may not feel cuts and blisters on their toes. These areas can become secondarily infected resulting in gangrenous digits that are often amputated (see image to left). This is part of the reason why all diabetics, especially poorly controlled diabetics, should see a foot specialist regularly.
Perhaps the most frightening complication of diabetes is the havoc it wreaks on the cardiovascular system. Diabetics have a much larger risk of heart attack and stroke.
Patients with diabetes are at risk for retinopathy, or damage to the retina of the eye. This can lead to blindness. Damage to the kidney’s filtering area known as the glomerulus can also occur; this can lead to chronic kidney disease. If severe enough, patients may need permanent dialysis or kidney transplantation.
Diagnosis
The diagnosis of diabetes is made by measuring the amount of sugar in the blood. There are three common ways to diagnose diabetes today. The first is by checking a molecule known as hemoglobin A1C. Diabetes can also be diagnosed with fasting blood sugar levels or by a glucose tolerance test. There are three possible results of these tests: normal, impaired (pre-diabetic), or diabetic.
Hemoglobin A1C levels are drawn from the blood. Two separate results above 6.5% indicate diabetes. Levels between 5.7 and 6.4% indicate pre-diabetes. Levels below 5.7% are normal.
"Fasting" blood glucose levels are usually taken in the morning before breakfast. It is considered "normal" if the blood glucose levels are between 60 and 100 mg/dL. Impaired is between 100 and 126 mg/dL. Diabetes is diagnosed if there are two fasting blood glucose levels greater than 126 mg/dL.
If fasting is not an option, or the patient ate breakfast, the clinician can do a "glucose tolerance test." In this test the patient drinks a liquid that is rich in sugar (75 grams of sugar is used for most adults). The blood sugar levels are then tested two hours later. If the blood sugar level at two hours is less than 140 mg/dL the test is "normal." If the level falls between 140 and 200 mg/dL the patient is impaired or pre-diabetic. And if the levels are greater than 200 mg/dL the diagnosis of diabetes is made.
Diagnosing Diabetes Mellitus
| Hemoglobin A1C | Fasting blood glucose levels | Glucose tolerance test levels at 2 hours after a 75g sugar load | |
|---|---|---|---|
| Normal | Less than 5.7% | < 100 mg/dL | < 140 mg/dL |
| Impaired | 5.7% to 6.4% | 100-126 mg/dL | 140-200 mg/dL |
| Diabetes | 6.5% or greater | > 126 mg/dL | > 200 mg/dL |
Heal Me Doctor
Treatment of diabetes involves either replacing insulin or improving insulin sensitivity.
Type 1 diabetics require insulin since their bodies no longer produce it. This is why type 1 diabetes was previously referred to as “insulin dependent diabetes”. Insulin as a treatment is discussed in a separate article.
(1) Replace insulin
(2) Improve insulin
sensitivity
There are numerous medicines used to control blood sugar levels in type two diabetics. One option is a class of medications known as the sulfonylureas. These medications work by increasing pancreatic production of insulin.
A second category of medications, known as the thiazolidinediones, improve the ability of insulin to act on peripheral tissues like fat, muscle, and liver.
The third category of medications, known as the biguanides, perform a similar function to the thiazolidinediones, and also inhibit liver cells from secreting stored sugar into the blood stream.
Regardless of which medications are used, the ultimate goal of medical therapy is to maintain the blood glucose levels within a specified range. Fasting blood glucose levels should ideally be kept between 90 and 130 mg/dL; post-meal glucose levels should ideally be less than 180 mg/dL. Maintaining glucose values within these ranges helps keep glycosylated hemoglobin (aka: hemoglobin A1C) levels below 7%, which decreases the risk of the diabetic complications.
In addition to replacing insulin, or increasing insulin sensitivity, patients with diabetes should also be treated for other co-morbid conditions. For example, statins are often started to control hyperlipidemia. Dialysis might be necessary to control severe kidney disease. Frequent foot and neurological exams are also necessary to prevent complications of diabetic neuropathy (ie: foot ulcers, gastric paresis, etc).
It is currently recommended that patients with diabetes have a blood pressure goal of 130/80 or better. In addition the LDL cholesterol level should be maintained below 100 mg/dL, HDL should be above 40 mg/dL, and triglycerides should be kept below 150 mg/dL.
The Replay…
Diabetes is classified as type 1 or type 2. In type 1 diabetes destruction of the pancreatic cells responsible for insulin secretion occurs through an auto-immune process. In type 2 diabetes, insulin sensitivity is lost by peripheral body tissues causing the pancreas to secrete more and more insulin until it “burns out”. Diagnosis is based off of blood sugar levels, usually measured when the patient is fasting (although other methods exist). Treatment is with insulin or insulin-sensitizing medications.
Just Keep Learning, Just Keep Learning (That’s a Dory Reference)…
- The Basics of Myocardial Infarction (Heart Attacks)
- Brain Boo Boos: Cerebrovascular Accidents (Stroke)
- Sugar Strip Down: Glycolysis and Energy Formation
- Hans Krebs and His Cycle: Oxidation and Energy
- The Electron Transport Chain in All It’s Glory
References and Resources
- Swanson A, Watrin K, Wilder L. Clinical Inquiries: How can we keep impaired glucose tolerance and impaired fasting glucose from progressing to diabetes? J Fam Pract. 2010 Sep;59(9):532-3.
- Judge EP, Phelan D, O’Shea D. Beyond statin therapy: a review of the management of residual risk in diabetes mellitus. J R Soc Med. 2010 Sep;103(9):357-62.
- Fowler GC, Vasudevan DA. Type 2 diabetes mellitus: managing hemoglobin A(1c) and beyond. South Med J. 2010 Sep;103(9):911-6.
- Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. Seventh Edition. Philadelphia: Elsevier Saunders, 2004.
- Le T, Bhushan V, Grimm L. First Aid for the USMLE Step 1. New York: McGraw Hill, 2009.
- Flynn JA. Oxford American Handbook of Clinical Medicine (Oxford American Handbooks of Medicine). First Edition. Oxford University Press, 2007.
- Champe PC. Lippincott’s Illustrated Reviews: Biochemistry. Second Edition. Lippincott-Ravens Publishers, 1992.
- American Diabetes Association. Standards of medical care in diabetes. Diabetes Care. 2004 Jan;27 Suppl 1:S15-35.