The pathology of amyotrophic lateral sclerosis (ALS) is not well known. There are familial forms, but they account for only 10% of cases. The familial form of amyotrophic lateral sclerosis is genetically heterogenous. This means that there are genes on different chromosomes that have been implicated in the disease process.
One of the most well studied is a gene known as superoxide dismutase (SOD) on chromosome 21. There are over 100 mutations of this gene. The defective protein that occurs from these mutations is believed to cause oxidative injury and/or toxic protein aggregates that damage motor neurons.
The remaining 90% of cases are sporadic. There is still debate about what causes most cases. Studies looking at cytoskeletal abnormalities, inflammation, and excitotoxicity from overactive glutamate stimulation have all been proposed as contributing factors.
The dying neurons are replaced with proliferating astrocytes in a process known as gliosis. This proliferation of glial cells leads to "glial scarring".
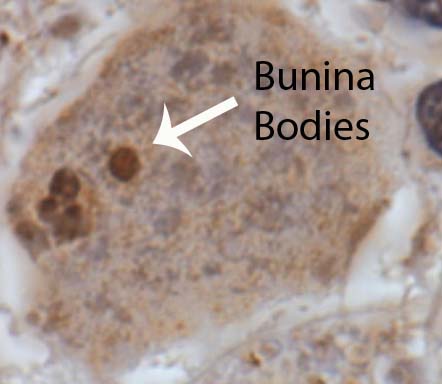
In addition, amyotrophic lateral sclerosis neurons have intracellular inclusions when viewed under the microscope. These inclusions are composed of different abnormal protein molecules that can be phosphorylated or ubiquinated. A specific type of inclusion known as a Bunina body is commonly seen.
Signs and Symptoms
Damage to the upper and lower motor neurons gives ALS its characteristic clinical presentation. Patients present with upper motor neuron findings: spastic weakness and hyper-reflexia (increased reflexes). Lower motor neuron death results in a flaccid weakness, atrophy of the muscles, muscular fasciculations (ie: abnormal twitching), and hypo-reflexia (decreased reflexes). Thus patients with ALS have a unique mix of both spastic and flaccid weakness, muscular atrophy, and hyper-reflexia (hyperactive reflexes usually predominate).
There are also many sub-categories of ALS. For example, "ALS Plus Syndrome" is characterized by the classic motor findings of amyotrophic lateral sclerosis plus dementia, Parkinsonism, autonomic nervous system instability, and sensory disturbances. Another sub-category known as bulbar palsy occurs when the symptoms of ALS predominately affect the face/cranial musculature. Cranial muscular weakness can cause difficulty swallowing (ie: dysphagia) and speaking (ie: dysarthria).
Diagnosis
Amyotrophic lateral sclerosis, for the most part, remains a clinical diagnosis. It is based upon finding the classic motor neuron findings on physical exam with symptoms that worsen over time. Unfortunately, there is still no laboratory test that definitively diagnoses the disease.
The El Escorial World Federation of Neurology criteria help guide the clinical diagnosis. In order to qualify for a diagnosis of amyotrophic lateral sclerosis a patient must meet the following criteria: evidence of lower motor neuron pathology on physical exam, electrophysiological, or neuropathological testing, and progressive worsening / spread of symptoms, as well as evidence of upper motor neuron disease on physical exam. In addition to the above, there must also be no evidence to support another possible explanatory diagnosis for the symptoms.
There are adjunctive tests that can support the diagnosis of amyotrophic lateral sclerosis. These include electromyography (EMG). EMG looks at the electrical activity produced in muscles. In amyotrophic lateral sclerosis, the EMG will generally show findings of denervation (ie: muscles that do not have nerves connecting to them). Fasciculation potentials are often seen on the EMG as well. Interestingly, nerve conduction studies are often normal in amyotrophic lateral sclerosis.
Finally, blood testing can help support the diagnosis. For example, creatine kinase (a marker of muscle damage) may be elevated, which is a result of denervation.
Other laboratory tests are often sent. Lyme disease antibodies, anti-nuclear antibodies, HIV, vitamin B12, thyroid function tests, calcium, and phosphorus are often sent to rule out other causes for the symptoms.
Treatment
Treatment for amyotrophic lateral sclerosis currently consists of a medication known as riluzole. This medication has been shown to improve survival rates.
In addition, supportive care is extremely important. Most patients eventually require tracheostomy and gastric tube placement in order to breath and eat. Motorized wheel chairs can help patients maintain some independence.
Regardless of treatment, the prognosis is extremely guarded. Most patients are deceased within 5 years of their diagnosis. Death is usually related to pulmonary complications like pneumonia.
Overview
Amyotrophic lateral sclerosis is a neurodegenerative disease that results in death of upper and lower motor neurons. Signs and symptoms are a bizarre mix of flaccid weakness with increased reflexes. Diagnosis is based on physical exam findings and symptoms that worsen over time. In addition, there should be no evidence that other causes are responsible. Treatment is with a medication known as riluzole, which has been shown to slow the progression of disease. Supportive treatment is extremely important for quality of life.
Learn More…
- Vitamin B12: Function and Disease
- Parkinson’s Disease: The Pathology of Movement
- The Direct Basal Ganglia Pathway
References and Resources
- Brooks BR. Managing amyotrophic lateral sclerosis: slowing disease progression and improving patient quality of life. Ann Neurol. 2009 Jan;65 Suppl 1:S17-23.
- Baehr M, Frotscher M. Duus’ Topical Diagnosis in Neurology: Anatomy, Physiology, Signs, Symptoms. Fourth Edition. Stuttgart: Thieme, 2005.
- Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. Seventh Edition. Philadelphia: Elsevier Saunders, 2004.
- Simon RP, Aminoff MJ, Greenberg DA. Clinical Neurology, Seventh Edition (LANGE Clinical Medicine). Seventh Edition. New York: McGraw Hill, 2009.
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006 Oct 5;52(1):39-59.
- Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. 1994 Jul;124 Suppl:96-107.