The notochord is a midline structure in the developing fetus that sends out various molecules (the most well known of which is “sonic hedgehog”). These molecules influence the development of the layers of embryonic cells that surround the notochord. One of these layers, the ectoderm, which is immediately behind (ie: posterior) the notochord eventually forms the brain and spinal cord. The mesoderm, which is immediately adjacent to the notochord forms the vertebral column and axial skeleton (amongst other things).
The purpose of the notochord is to ensure that each layer of tissue forms what it is supposed to. Once this occurs, the notochord ultimately becomes the nucleus pulposus of the intervertebral discs.
In some people, nests of cells that composed the fetal notochord remain (unnaturally) after birth. These collections of cells are known formally as ecchordoses physaliphora (try saying that 5 times fast). These cells can divide and turn into a slow growing tumor… What is that tumor called? You guessed it! A chordoma!
Chordomas are slow growing tumors that are most commonly located at the ends of the vertebral column. The most common place to see them is in the sacrococcygeal region, followed in frequency by a bone known as the clivus at the base of the skull. However, chordomas can occur anywhere along the vertebral column. The reason they occur most frequently at the "ends" of the vertebral column (ie: skull base and sacrum) is because these are the last areas to fuse in-utero.
Additionally, since the notochord is a midline structure in the fetus, chordomas are almost always midline in location.
Microscopically chordomas contain large polygonal shaped cells embedded in a mesh of long repetitive sugar and nitrogen containing molecules known as mucopolysaccharides.
– Arise from notochord cells
– S-100 positive
– Cytokeratin positive
– Polygonal cells
– Slow growing
– Midline location
– Sacrum and clivus most
common locations
– Worse prognosis than
chondrosarcomas
Distinguishing chondrosarcomas from chordomas is possible with immunocytologic staining techniques. Chondromas and chondrosarcomas stain positive for a protein known as S-100. S-100 proteins have numerous intracellular functions and are commonly present in cells such as adipocytes (fat cells), chondrocytes (cartilage forming cells), melanocytes (pigment producing cells), and Schwann cells, amongst others.
So if chondrosarcomas and chordomas can look alike, and both stain positive for S-100, how the heck do we distinguish between the two? Using another molecule known as cytokeratin! Cytokeratin is a molecule that forms part of the intracellular frame for many cells. It is present in chordomas, but not in chondrosarcomas.
On to the Clinic I Say…
Chordomas are slow growing tumors and will usually start to cause symptoms in mid-adulthood. Symptoms are based on the location of the tumor.
If the tumor is located in the sacrum or coccyx then pain is the most frequent presenting symptom. If undiscovered this may progress to bowel and bladder problems as the tumor slowly envelopes the sacral nerves that go to the bowel and bladder. Additionally, patients may present with radicular symptoms such as numbness, tingling, or sharp pain in the distribution of the sacral nerves.
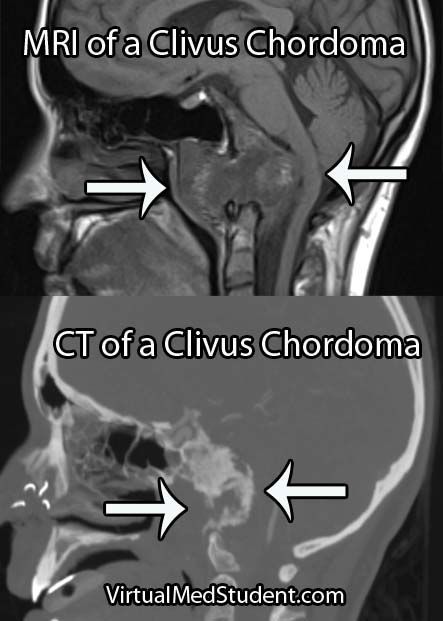
Diagnose Me McCoy
Diagnosis of chordoma can only be officially made by looking at the specimen under a microscope. However, imaging studies with x-rays, CT scans, and MRI imaging can support the diagnosis. Imaging studies will typically show a midline lytic lesion centered in the bone. Invasion of adjacent anatomical structures can occur, but is a late manifestation of the disease course.
Treating These Ugly Tumors
The gold-standard treatment for chordomas is en-bloc surgical resection with wide margins followed by radiation therapy. Complete resection is difficult, if not impossible to achieve in the skull base, but may be possible in the spine and/or sacrum with very skilled surgical teams.
Without an en-bloc surgical resection the risk of tumor re-growth and recurrence is very high. If you are planning to biopsy of a sacral lesion you should mark the biopsy tract with methylene blue so that the tract can also be resected during surgery as tumor cells can seed the tract as the needle is being pulled out.
Let’s Remix This Overview
Chordomas are slow growing, but malignant tumors that arise from notochord cells that fail to regress after birth. They are most frequently found in the sacrum and clivus (one of the bones constituing . Diagnosis is made with CT, MRI, and x-rays, as well as via tissue diagnosis at the time of surgical removal. Chordomas stain positive for S-100 and cytokeratin proteins, which helps distinguish them from chondrosarcomas that only stain positive for S-100. Symptoms are based on where the lesion is located (skull base or spine). Treatment is surgery followed by radiation therapy.
Just Warming Up…
- Acoustic neuroma (vestibular schwannoma)
- Cavernous malformation (cavernoma)
- Ependymoma
- Glioblastoma
- Hemangioblastoma
- Hemangiopericytoma
- Meningioma
- Pituitary adenoma
- Colloid cyst of the third ventricle
Want to Research More About Chordomas?
- Yeom KW, Lober RM, Mobley BC, et al. Diffusion-Weighted MRI: Distinction of Skull Base Chordoma From Chondrosarcoma. AJNR Am J Neuroradiol. 2012 Nov 1.
- Walcott BP, Nahed BV, Mohyeldin A, et al. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012 Feb;13(2):e69-76.
- Almefty K, Pravdenkova S, Colli BO, et al. Chordoma and chondrosarcoma: similar, but quite different, skull base tumors. Cancer. 2007 Dec 1;110(11):2457-67.
- Cho YH, Kim JH, Khang SK, et al. Chordomas and chondrosarcomas of the skull base: comparative analysis of clinical results in 30 patients. Neurosurg Rev. 2008 Jan;31(1):35-43; discussion 43.
- Yoneoka Y, Tsumanuma I, Fukuda M, et al. Cranial base chordoma–long term outcome and review of the literature. Acta Neurochir (Wien). 2008 Aug;150(8):773-8; discussion 778.
- Amichetti M, Cianchetti M, Amelio D, et al. Proton therapy in chordoma of the base of the skull: a systematic review. Neurosurg Rev. 2009 Oct;32(4):403-16.
- Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease
. Seventh Edition. Philadelphia: Elsevier Saunders, 2004.