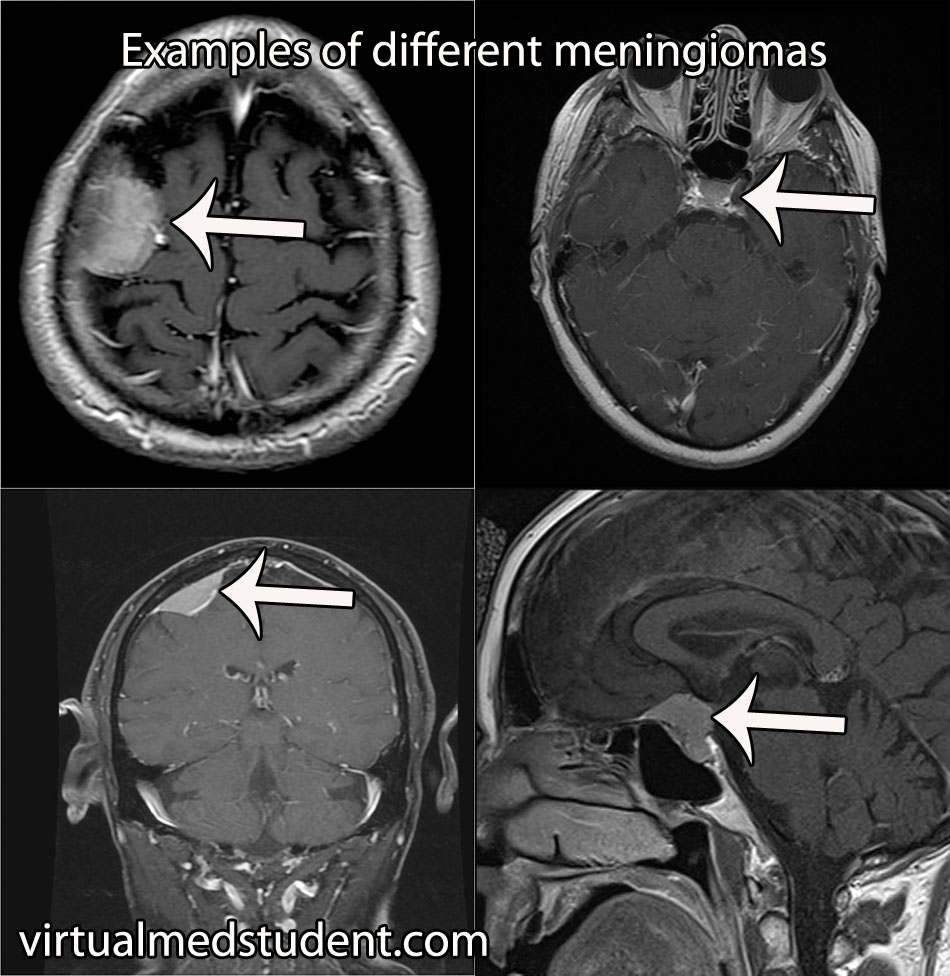
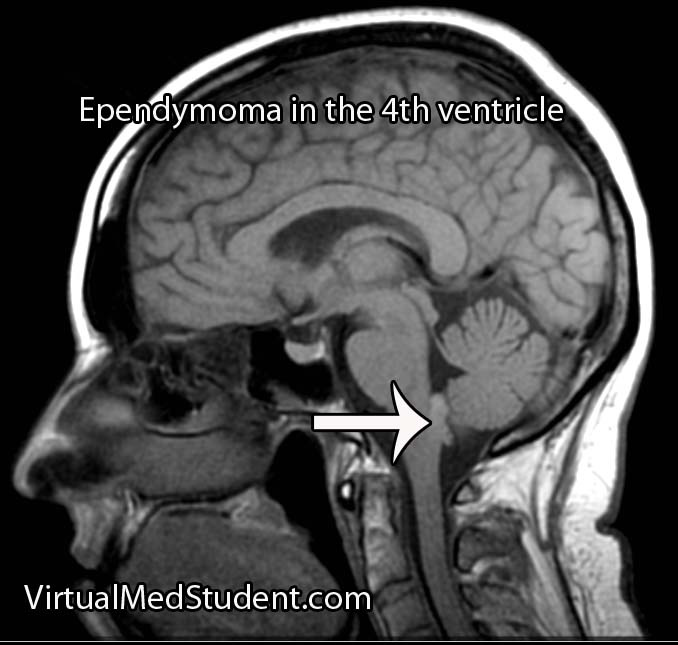
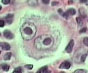
Hemangioblastomas can be solid or cystic in appearance. However, all hemangioblastomas have capillary networks that are lined by endothelial cells. Interspersed between the capillaries are pericytes and stromal cells with a polygonal appearance.
The arrangement of these various cell types can occur in three different architectures: juvenile, clear cell, or transitional. Each architecture has a specific ratio of capillaries to stroma (connective tissue), with the juvenile type having the greatest amount of capillary tissue, and the clear cell type having the greatest amount of stromal tissue.
About a quarter of cases are related to a genetic disorder known as von Hippel-Lindau syndrome. von Hippel-Lindau syndrome is the result of a genetic mutation in the VHL gene on chromosome 3. The protein product of this gene is a tumor suppressor; when mutated it is unable to suppress the abnormal growth of tumor cells. As a result, patients with von Hippel-Lindau syndrome develop hemangioblastomas of the brain and retina, renal cell carcinoma, and other tumor types.
Hemangioblastomas are most commonly found in the cerebellum, but on occassion will affect the cervical spinal cord and brainstem. They are most commonly seen in males starting at around 20 years of age.
Signs and Symptoms
Patients with cerebellar hemangioblastomas present with numerous signs and symptoms. Many patients will complain of headache, nausea, and vomiting. This is often due to the tumor compressing the cerebral aqueduct, which causes cerebrospinal fluid to “back up” in the brain leading to hydrocephalus and increased pressure inside the head.
In addition, many people with hemangioblastomas will have evidence of cerebellar dysfunction on physical exam. These signs include ataxia (ie: wobbly gait), dysmetria (ie: uncoordinated movements of the limbs), and dysdiadochokinesia (ie: difficulty repeating rapid alternating movements).
If the tumor is present in the spinal cord, symptoms may include weakness, spasticity, numbness, or other sensory changes.
Interestingly, hemangioblastomas can secrete an analogue of the hormone erythropoietin. This hormone causes bone marrow to pump out more red blood cells. As a result, some patients may have an increased number of red blood cells; this is known as polycythemia.
Diagnosis
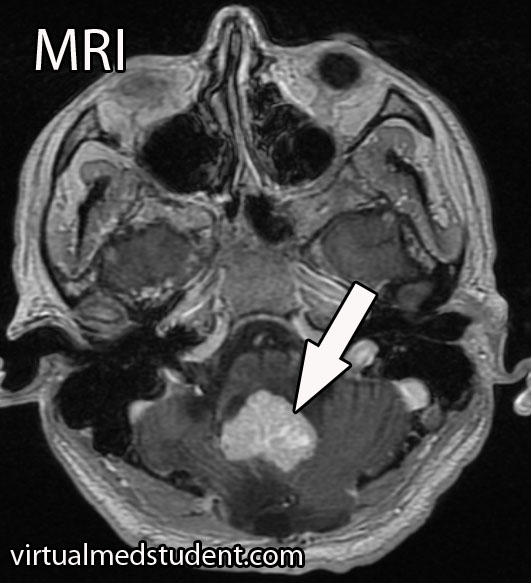
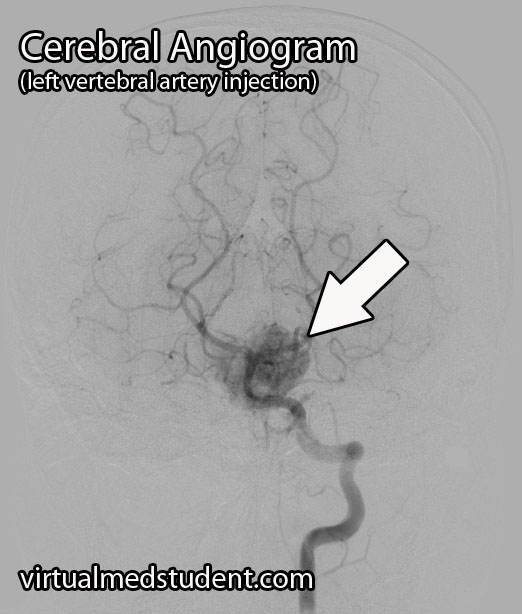
A tumor located in the cerebellum of an adult with certain characteristics on CT, MRI, and cerebral angiogram can make the diagnosis of hemangioblastoma likely.
However, the final diagnosis can only be made by looking at a sample of the tumor under the pathology microscope.
Treating These Bastards
Definitive treatment is surgical resection. However, hemangioblastomas can be highly vascular, which means they tend to bleed like stink! Therefore, preoperative embolization by an interventional neuro-radiologist can decrease the amount of bleeding that occurs during surgery.
Radiation therapy is also sometimes used as an adjunctive treatment. It may help slow the growth of the tumor, but will not cure it. Radiation therapy can be used in patients who are unable to undergo surgical resection, or if the tumor is inaccessible via traditional surgical means.
Recap It All…
Hemangioblastomas are highly vascular, but benign central nervous system tumors of undetermined origin. They are associated with von Hippel-Lindau syndrome. Signs and symptoms include headache, nausea, and vomiting, as well as cerebellar dysfunction. Diagnosis is based on imaging and pathological findings at the time of surgical resection. Surgery is the treatment of choice, although embolization and radiation therapy may also be used as an adjunct.
More Interesting Neurological Problems…
- Hydrocephalus: Greek for Water Head
- Cerebral Arteriovenous Malformations: A Disease of Eloquence
- Vein of Galen Malformations: A Misnomer of Sorts
- Cerebral Cavernous Malformations: Leaky Vessels
Some More Expert Ideas Below
- Ho VB, Smirniotopoulos JG, Murphy FM, et al. Radiologic-pathologic correlation: hemangioblastoma. AJNR Am J Neuroradiol. 1992 Sep-Oct;13(5):1343-52.
- Silver ML, Hennigar GJ. Cerebellar hemangioma (hemangioblastoma); a clinicopathological review of 40 cases. J. Neurosurg. 1952 Sep;9(5):484-94.
- Kaelin WG Jr. The von hippel-lindau tumor suppressor protein: an update. Methods Enzymol. 2007;435:371-83.
- Gläsker S. Central nervous system manifestations in VHL: genetics, pathology and clinical phenotypic features. Fam Cancer. 2005;4(1):37-42.
- Gläsker S, Van Velthoven V. Risk of hemorrhage in hemangioblastomas of the central nervous system. Neurosurgery. 2005 Jul;57(1):71-6; discussion 71-6.
- Moss JM, Choi CY, Adler JR Jr, et al. Stereotactic radiosurgical treatment of cranial and spinal hemangioblastomas. Neurosurgery. 2009 Jul;65(1):79-85; discussion 85.