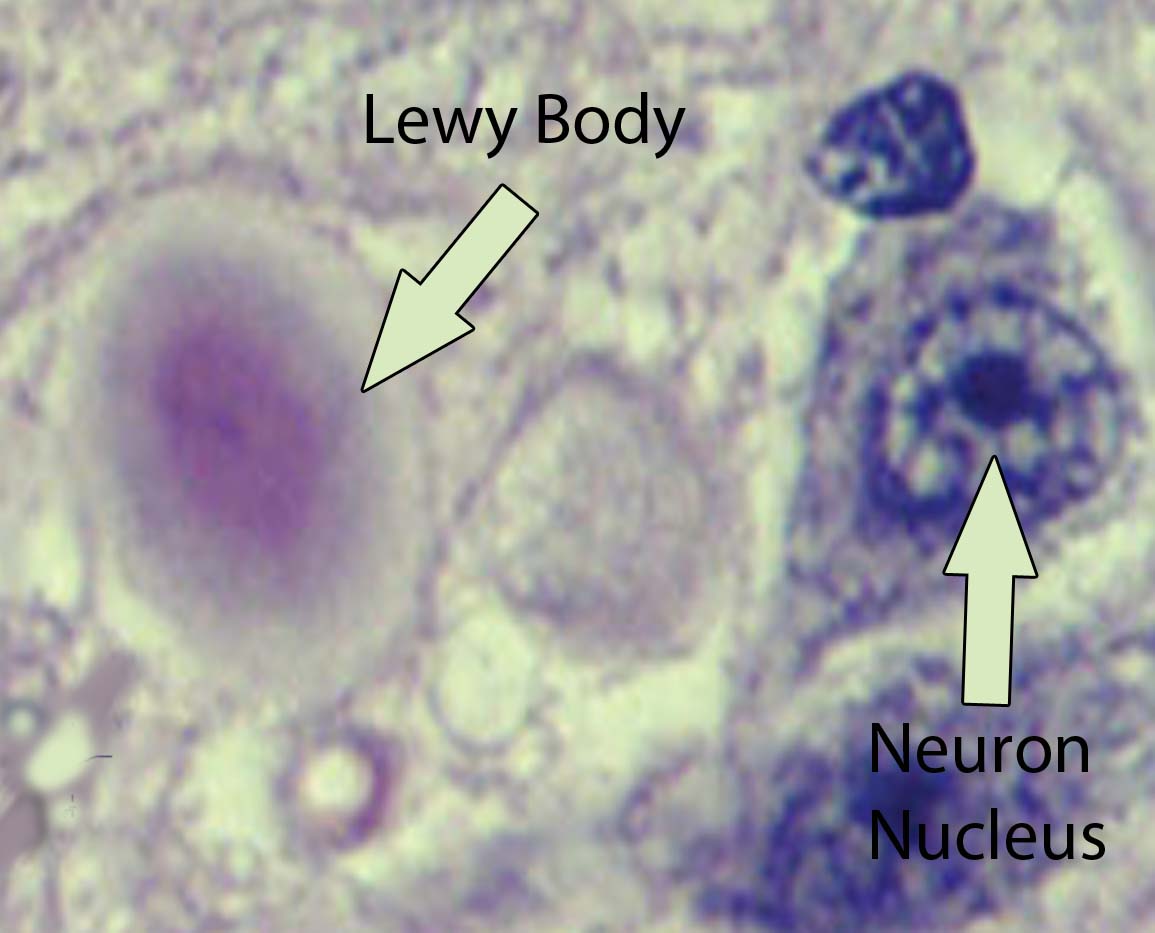
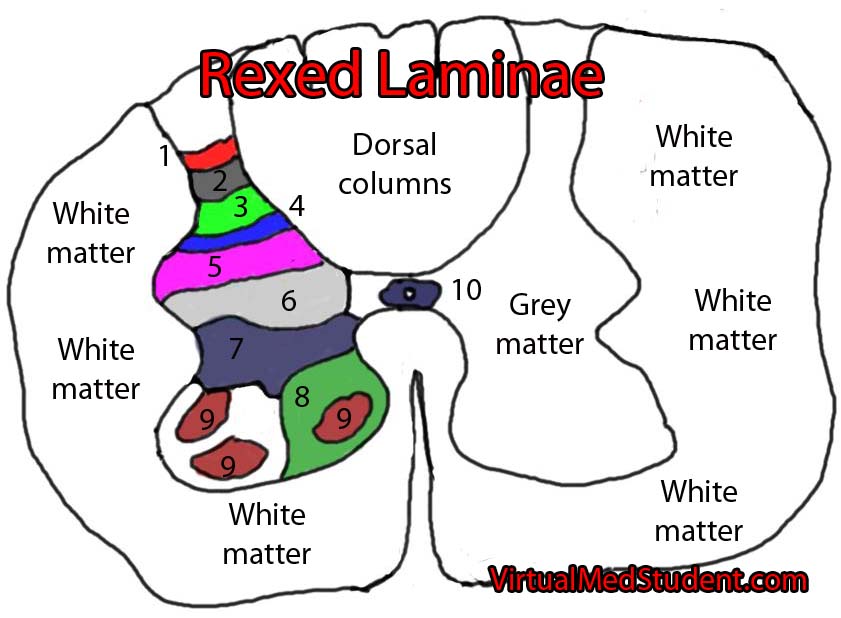
The Rexed laminae are a system for organizing the neurons of the spinal cord (they were actually designed to torture medical students, I joke of course, sort of…). They are named after Dr. Bror Rexed who was a Swedish neuroscientist.
There are ten Rexed laminae and they follow a topographic organization, with the lower numbers being towards the back of the spinal cord (ie: posterior) and the higher numbers being towards the front of the spinal cord (ie: anterior).
The Rexed laminae are not strictly organized based on anatomical location, but are actually based on the types, and functions, of the neurons in each laminae. Let’s discuss each of the ten layers in more detail (let the fun begin!)…
Layer one contains neurons that receive pain and temperature information from the body and limbs via the axons coming from the dorsal root ganglia. The neurons in layer one then pass this information along to the brain via the spinothalamic tract on the opposite side of the cord.
Layer two, which is also known as the substantia gelatinosa, gets information from the spinothalamic tract as well as the dorsal columns. The spinothalamic tract relays information about painful stimuli and the dorsal columns relay information about non-painful stimuli.
Therefore, the neurons in layer two receive information about both painful and non-painful stimuli. These neurons then send information to Rexed laminae three and four. The neurons in these laminae then pass the sensory information to the brain where it is further interpreted. Interestingly, there are large amounts of opiate (ie: think morphine or heroin) responsive neurons in laminae two of the spinal cord.
The nucleus proprius, which constitutes layers three and four, receives information from the body about touch and proprioception (hence the name “proprius”). It then relays this information to numerous areas in the brainstem, brain, and other Rexed laminae for further processing.
Nobody knows exactly what the heck layer five does, but it receives information from a wide variety of sources including pain sensation from the bodies’ organs, as well as information about movement from the brain (via the corticospinal tracts) and brainstem (via the rubrospinal tracts).
The sixth layer can be divided into two sections: medial and lateral. The medial layer (ie: towards the middle of the spinal cord) gets input from muscle spindles. Muscle spindles (by way of type Ia fibers) tell the spinal cord how much a given muscle is being stretched. The neurons in the medial layer act as messengers for this information.
So what is the purpose of the sixth layer? The neurons in this layer send information to two places: the cerebellum (via the ventral spinocerebellar tracts) and motor neurons in the anterior horn of the spinal cord.
The cerebellum interprets the information and modulates movement and muscle tone accordingly. The direct communication between the neurons in Rexed laminae six and the motor neurons in the anterior horns are responsible for spinal reflexes.
Layer seven is most prominent between C8 and L2. This prominence is known as the dorsal nucleus of Clarke. The neurons in Clarke’s nucleus receive lower extremity position and sensory information and then pass that information to the cerebellum via the dorsal spinocerebellar tract.
Layer seven also receives and sends information from, and to, the bodies’ organs. The sympathetic and parasympathetic autonomic system have their pre-ganglionic neurons in Rexed laminae seven. These neurons are responsible for the fight or flight response (sympathetic) and rest and digest (parasympathetic) functions of the bodies’ organs.
Neurons in layer eight obtain information from the reticulospinal and vestibulospinal tracts. The reticulospinal tract is important in maintaing the tone of muscles that flex joints. The vestibulospinal system helps maintain the tone of muscles that are important in extending joints.
So what do the neurons in layer eight do? They take competing information from the reticulospinal and vestibulospinal tracts and help modulate movement and muscular tone. Pretty cool, huh?
Finally an easy layer! This layer contains the α, β, and γ motor neurons of the cord. Simply stated, these neurons send impulses to muscles leading to movement and its’ modulation. The more complex story is that motor neurons in layer nine are influenced by numerous inputs from other Rexed laminae, as well as by descending information coming from the brain.
The neurons in layer nine are topographically organized based on what type of muscle (flexor or extender) they control, as well as where in the body that muscle is located (axial or limb).
Neurons that are located towards the center of the cord control axial muscles and those located towards the periphery of the cord control the muscles of the limbs. In similar fashion, neurons that control muscles that extend joints are located towards the front of the layer (ie: anterior), and those that flex joints are located towards the back of layer nine (ie: posterior).
Like layer five, no one really knows what layer ten actually does. We do know that it is composed of neurons that surround the fluid filled central canal of the spinal cord like a donut. Hungry yet???
Overview
The Rexed laminae are layers of neurons within the spinal cord that perform specific functions. In general, neurons in the laminae towards the back of the cord (ie: laminaes one, two, three, four, and five) are predominately involved in interpreting and relaying sensory information from the body to the brain. On the other hand, neurons in the laminae towards the front of the cord (ie: laminaes seven, eight, and nine) are involved primarily in executing movement and controlling the functions of the bodies organs.
References and Resources
- Kitahata L, Kosaka Y, Taub A, et al. Lamina-specific suppression of dorsal-horn unit activity by morphine sulfate. Anesthesiology, V41, issue 1, 1974.
- Anamizu Y, Seichi A, Tsuzuki N, et al. Age-related changes in histogram pattern of anterior horn cells in human cervical spinal cord. Neuropathology. 2006 Dec;26(6):533-9.
- Stephens B, Guiloff RJ, Navarrete R, et al. Widespread loss of neuronal populations in the spinal ventral horn in sporadic motor neuron disease. A morphometric study. J Neurol Sci. 2006 May 15;244(1-2):41-58.
- Baehr M, Frotscher M. Duus’ Topical Diagnosis in Neurology: Anatomy, Physiology, Signs, Symptoms. Fourth Edition. Stuttgart: Thieme, 2005.
- Neuroscience. Fourth Edition. Sinauer Associates, Inc., 2007.
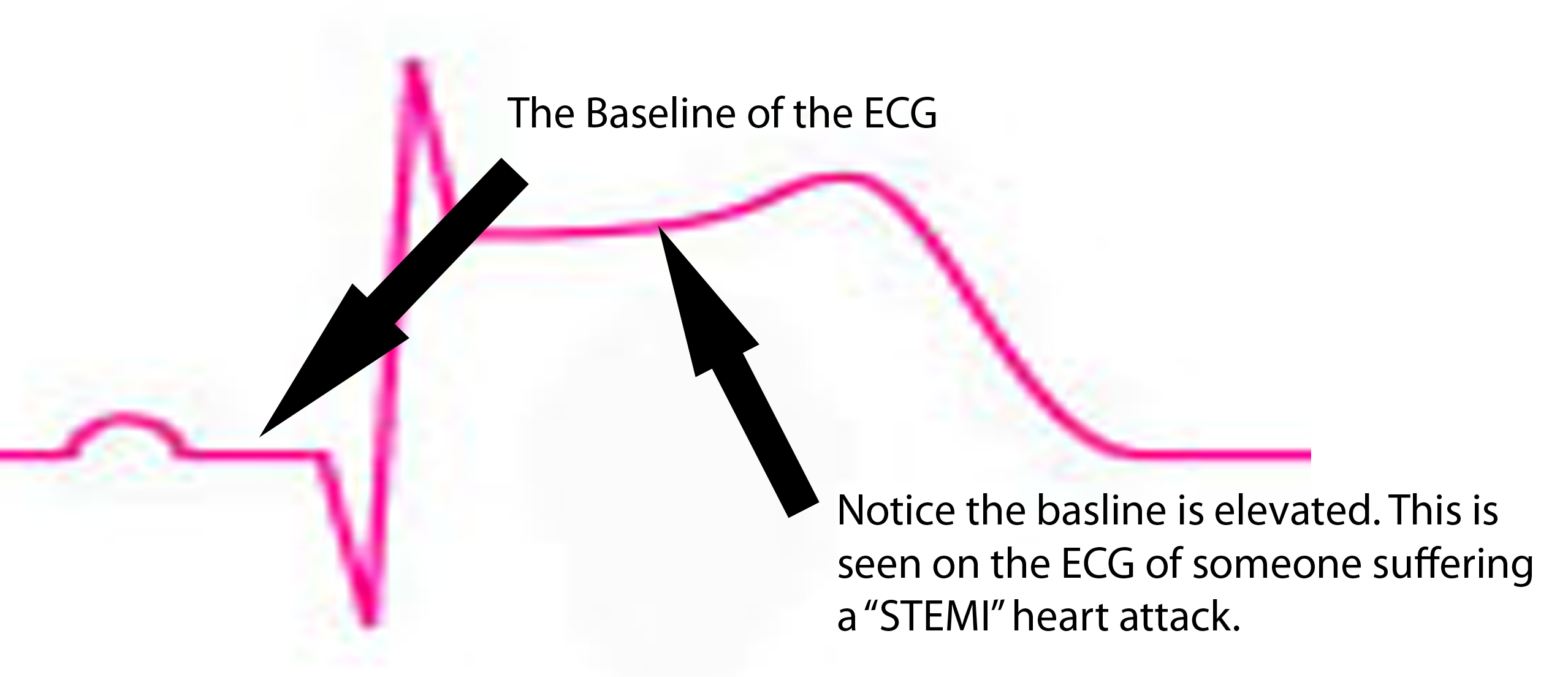
 ST segment is depressed below baseline.
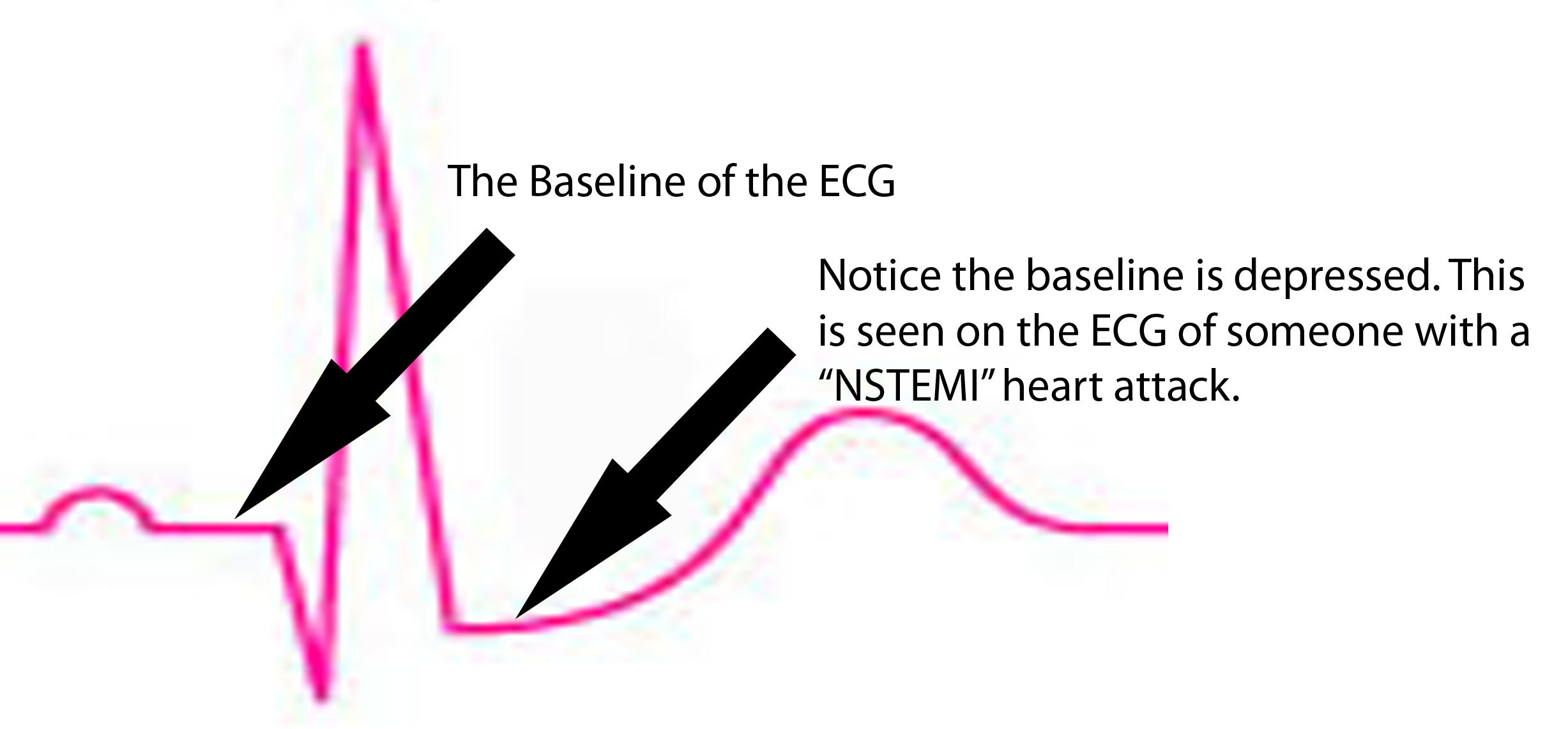
ST segment is depressed below baseline.