The cardiac output (CO) measures how much blood the heart pumps per minute. It is directly related to the stroke volume (SV) and heart rate (HR). The stroke volume is the amount of blood in the left ventricle of the heart just before it contracts. The cardiac output is calculated by multiplying the heart rate by the stroke volume (CO = HR x SV).
The cardiac output is related to Ohm’s law, which in electrical terms states that the change in voltage of a circuit is equal to the flow of current, I, multiplied by the resistance, R, of the circuit (ΔV = I x R).
Like electrical current in a circuit, blood flows in a circular pathway from the left ventricle, through the body, where it eventually ends up in the right ventricle. We can change Ohm’s law to govern hemodynamics by stating that the change in voltage is equivalent to the change in pressure between the aorta and right atrium (mean arterial pressure and central venous pressures, respectively), flow of current is equal to the amount of blood pumped per unit time (ie: cardiac output), and resistance is equal to the resistance the blood sees as it travels through the vessels of the body (aka: the systemic vascular resistance).
Therefore, if we re-write Ohm’s law for the hemodynamics of cardiac output we get: central venous pressure (CVP, measured in mmHg) subtracted from the mean arterial pressure (MAP, measured in mmHg) is equal to cardiac output (CO, measured in liters/minute) multiplied by the systemic vascular resistance (SVR, measured in dynes-s / cm5), or:
(MAP – CVP) = (CO x SVR) / 80
(ΔV = I x R)
Doing a simple mathematical rearrangement of the equation above we get:
CO = [(MAP – CVP) / SVR] x 80
*** The 80 in the equation is a conversion factor to convert Wood to metric units ***.
In essence, the cardiac output is directly proportional to the difference in blood pressure between the left (arterial side) and right (venous side) sides of the body, and inversely proportional to the vascular resistance. In other words, if the pressure difference between the arterial and venous side of the body decreases the cardiac output will fall. Along the same lines, if the systemic vascular resistance increases then cardiac output will decrease.
Clinical Measurements
How do you clinically measure the components that constitute the cardiac output?
The most reliable, but invasive way is to use a pulmonary artery catheter, also known as a "Swan Ganz" catheter. The catheter is inserted into the pulmonary artery and advanced until it “wedges” in a small branch of the pulmonary arterial tree. From there a balloon in the tip of the catheter is inflated to keep it in place. At this location the tip of the catheter is effectively measuring the pressure of blood in the left atrium (ie: the "pulmonary artery wedge pressure"). Using the catheter, and various methods such as the thermodilution technique, the cardiac output and/or stroke volume can be measured directly.
Unfortunately, the use of Swan-Ganz catheters are fraught with serious problems such as arrhythmias and infection. Therefore, other less invasive techniques such as echocardiography with doppler (ie: an ultrasound of the heart) can be used to estimate cardiac output and stroke volume.
The other elements of cardiac output can also be measured clinically. The mean arterial pressure is calculated from the systolic and diastolic blood pressures, which are measured from a cuff or arterial catheter. Central venous pressure can be measured with a central venous catheter (ie: a "central line").
Role in Disease
A normal cardiac output is roughly 5.5 L/min for an average sized male and 5.0 L/min for an average sized female. However, in individuals with diseased heart muscle the pumping ability of the heart is reduced. The result is a decrease in cardiac output.
So what’s the big deal? A fall in cardiac output means that the body sees less oxygenated blood per minute. Every organ in the body requires oxygen to function properly. If cardiac output is decreased the organs may not receive enough oxygen to do their job. In super severe cases, when cardiac output approaches zero, cardiogenic shock occurs. If no oxygen reaches the organs multi-organ failure occurs and the patient may die.
Overview
Cardiac output is related to the stroke volume, heart rate, systemic vascular resistance, and difference in pressure between the arterial and venous sides of the body. When cardiac output drops the bodies’ organs receive less oxygen per unit time. In severe cases this may lead to organ death.
Related Articles
- Dilated Cardiomyopathy: Poor Pump, S3, and Crackles
- Systemic Vascular Resistance: Radius, Length, Viscosity
- AV Heart Block: PR, QRS, and Mobitz
References and Resources
- Alhashemi JA, Cecconi M, della Rocca G, et al. Minimally invasive monitoring of cardiac output in the cardiac surgery intensive care unit. Curr Heart Fail Rep. 2010 Sep;7(3):116-24.
- Noritomi DT, Vieira ML, Mohovic T, et al. Echocardiography for hemodynamic evaluation in the intensive care unit. Shock. 2010 Sep;34 Suppl 1:59-62.
- Magder S. Fluid status and fluid responsiveness. Curr Opin Crit Care. 2010 Aug;16(4):289-96.
- Swann DG. The utility of pulmonary artery catheterization. Br J Anaesth. 2000 Oct;85(4):501-3.
- Rodriguez R, Hern HG Jr. An approach to critically ill patients. West J Med. 2001 Dec;175(6):392-5.
- Lilly LS, et al. Pathophysiology of Heart Disease: A Collaborative Project of Medical Students and Faculty
. Fourth Edition. Lippincott Williams and Wilkins, 2006.
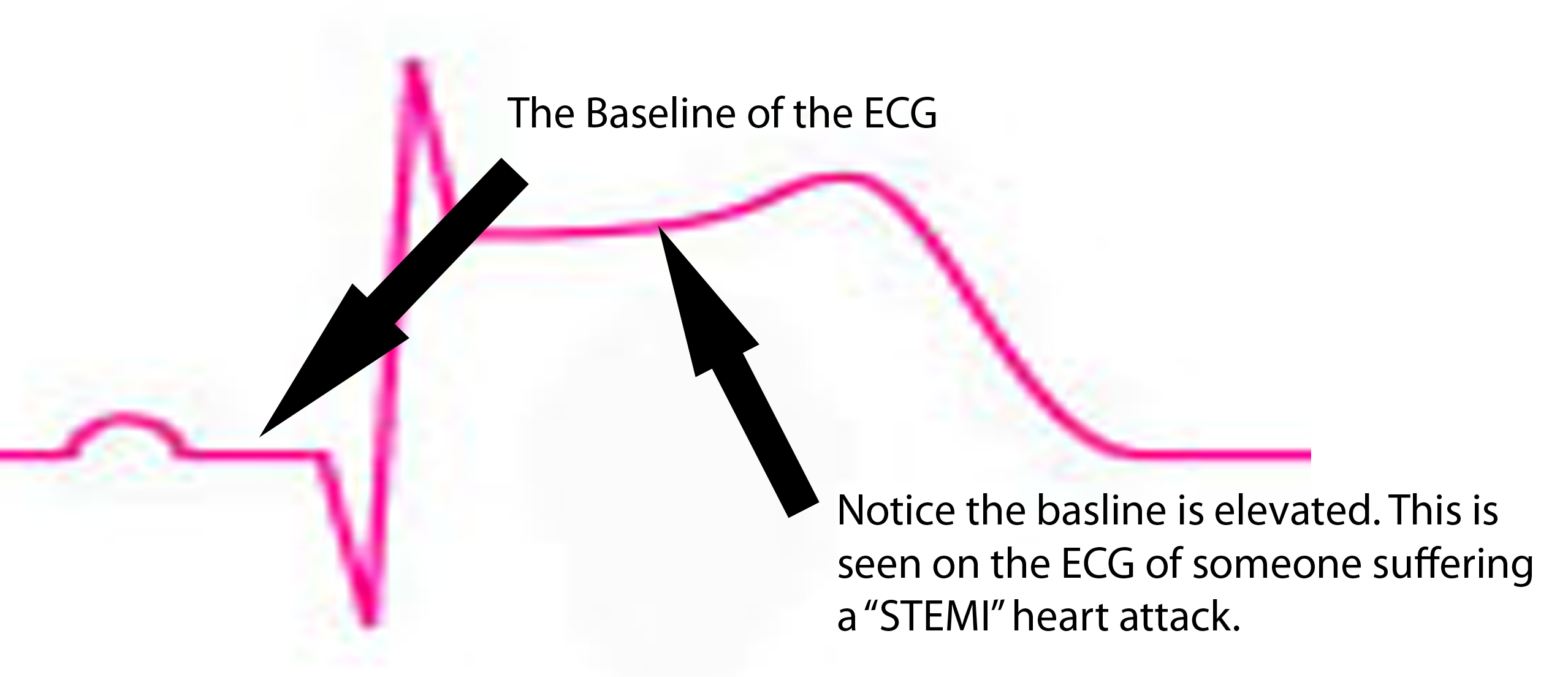
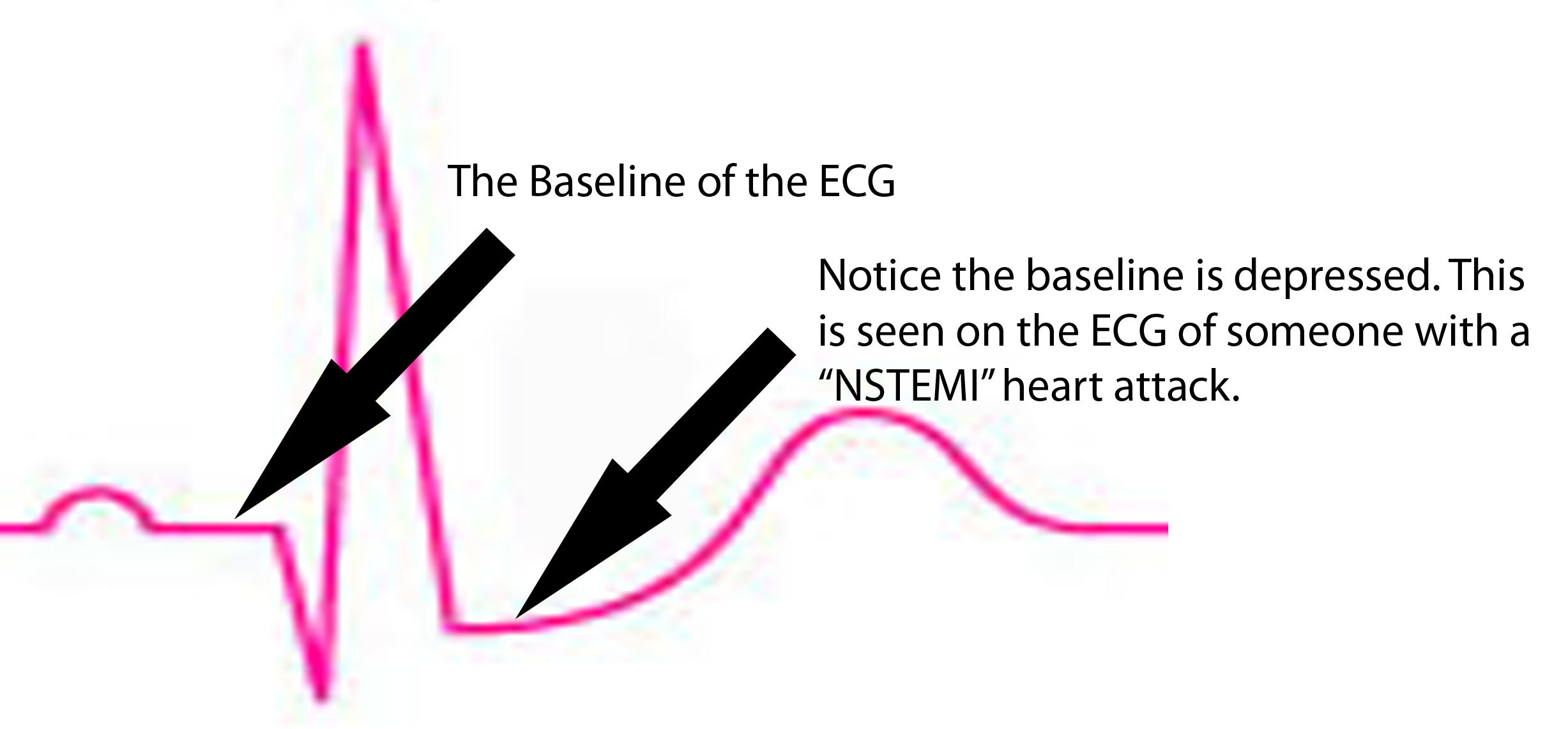 ST segment is depressed below baseline.
ST segment is depressed below baseline.