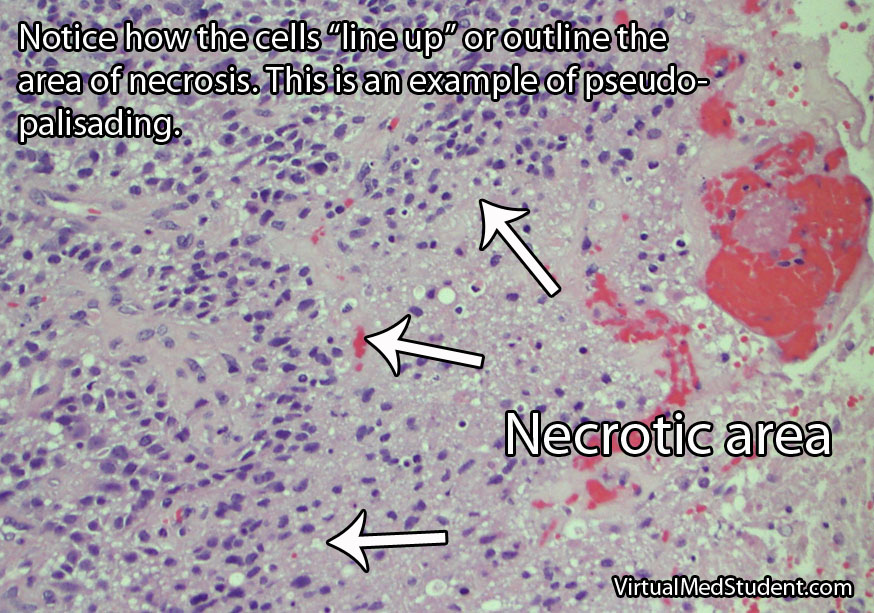
When viewed under the light microscope, meningiomas can have many different appearances. The most common type is a dense sheet-like formation of cells interspersed with closely packed blood vessels. Sometimes the sheets of cells can be separated by connective tissue.
Frequently, meningiomas will have calcium deposits in them known as “psammoma bodies”. Uncommonly, meningioma cells may take on a malignant appearance characterized by increased cellular division (ie: “mitotic figures”) and invasion of the tumor cells into surrounding brain or bone.
Meningiomas test positive for epithelial membrane antigen (EMA) and vimentin (a marker of connective tissue). Ki-67 (a marker of proliferation) can be elevated in more aggressively behaving tumors.
The most common genetic abnormality seen in patients with meningiomas is found on chromosome 22. If present, a mutation in the NF2 gene on this chromosome causes type 2 neurofibromatosis. This disease predisposes individuals to developing multiple tumor types including meningiomas. Other less common genetic abnormalities can be seen on other chromosomes as well.
Signs and Symptoms
Due to their slow growing and benign nature, many meningiomas cause no symptoms. However, if they become too large or start to compress adjacent brain tissue they can cause headache, seizures, confusion, or visual problems. Spinal cord compression can result in myelopathy.
The most common location for a meningioma is in between the two hemispheres of the brain – the so called “parasagittal” location. Parasagittal meningiomas near the portion of the brain responsible for muscle movements may cause weakness of the opposite leg.
Diagnosis
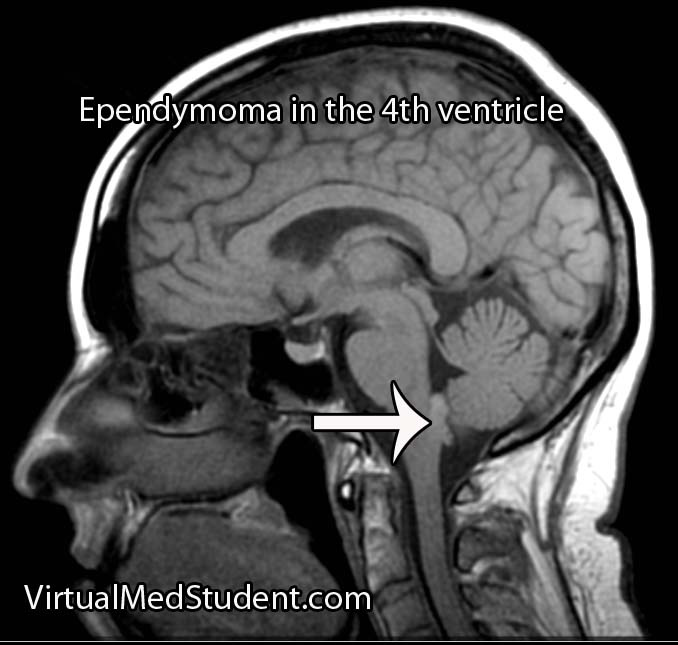
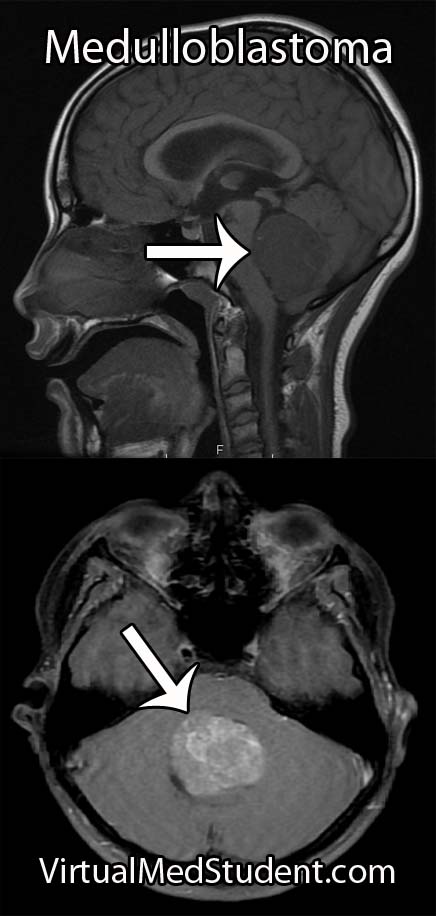
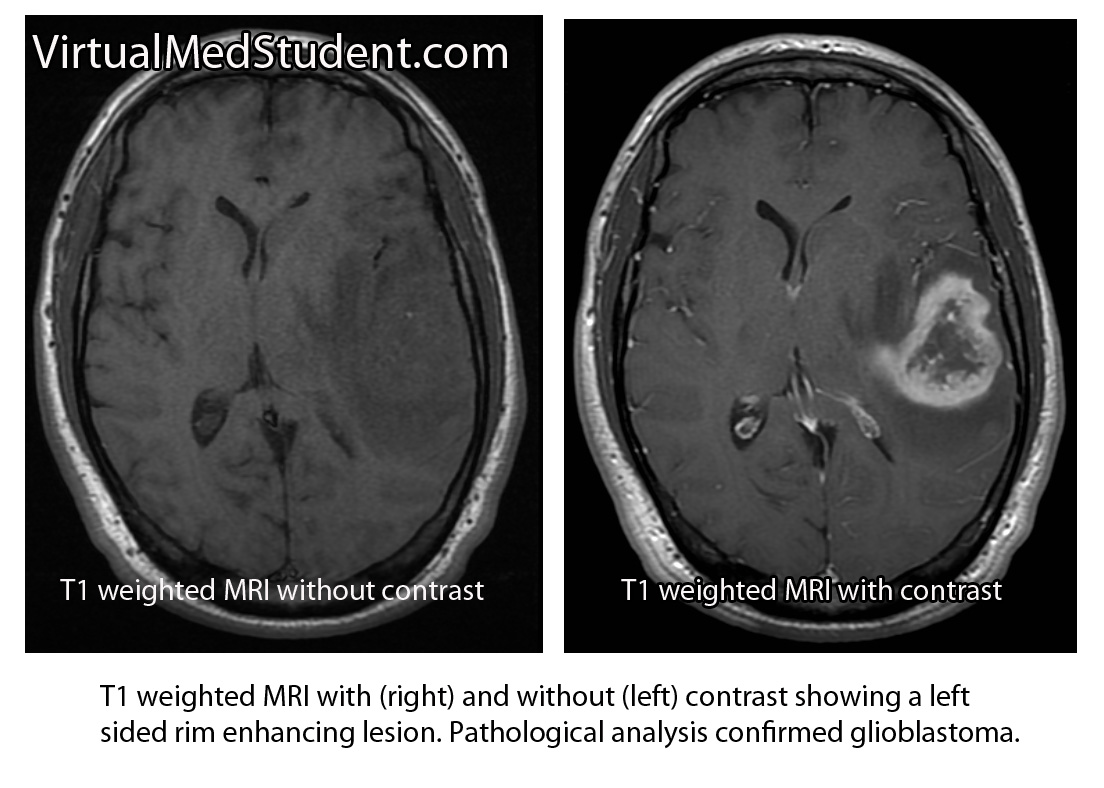
Diagnosis of meningioma can reliably be made on characteristic findings seen on CT or MRI scans. Interestingly, many meningiomas are found incidentally when a CT or MRI is done for other reasons.
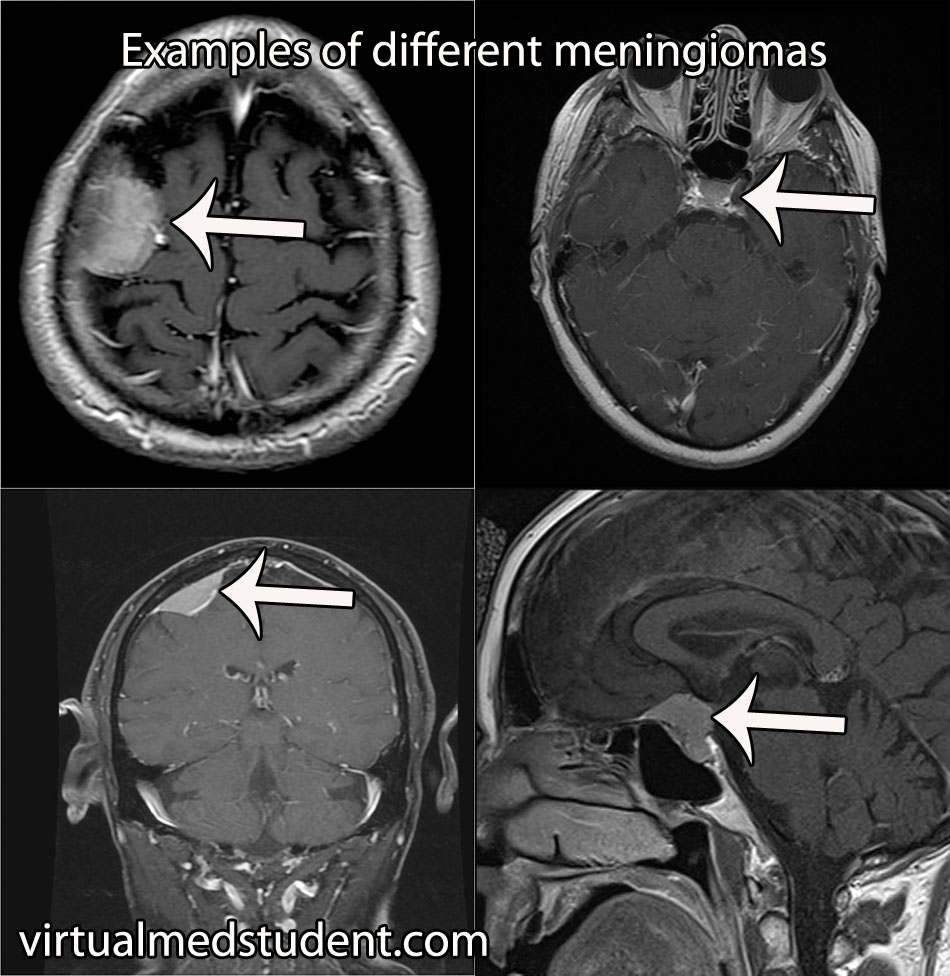
However, like any other tumor, meningiomas can only be truly diagnosed once a specimen is sent to the pathology lab for analysis. Pathologists can reliably make the diagnosis based on typical histological features.
Meningiomas must be distinguished from a more malignant tumor known as a hemangiopericytoma. Hemangiopericytomas can look similar to meningiomas on imaging studies.
Dish Me Up Some Treatment Sir
Many meningiomas can be watched over time with repeat imaging studies; this is especially true if they are small and not causing neurological signs or symptoms.
On the other hand, large or symptomatic meningiomas require surgical resection. Many meningiomas can be removed completely. However, some meningiomas may be near vital structures such as the carotid artery, cranial nerves, or venous draining systems of the brain where complete surgical removal may be very difficult without causing significant neurologic impairment. In these cases the tumor is debulked as much as possible. The residual tumor can be followed or irradiated depending on the grade of meningioma.
Residual tumor after incomplete surgical resection, or meningiomas in difficult to access locations are candidates for radiation therapy. Many studies have shown long term growth control rates.
Overview
Meningiomas are considered “benign” tumors of the brain. They arise from arachnoid cap cells, which are located in a layer of the meninges (ie: the covering of the brain) known as the arachnoid. Symptoms include headache, weakness, vision problems, paresthesias (ie: abnormal sensations), amongst many other possible symptoms. Diagnosis can be made reliably from imaging studies such as CT or MRI. If symptomatic, or large, treatment is surgical resection. Small asymptomatic meningiomas can be managed with repeat imaging to assess for growth over time.
Related Articles
- Colloid cyst of the third ventricle
- Acoustic neuroma
- Hemangioblastoma
- Cavernous malformations
- Hemangiopericytoma
Curated References for Your Pleasure…
- Ragel BT, Jensen RL. Aberrant signaling pathways in meningiomas. J Neurooncol. 2010 Sep;99(3):315-24. Epub 2010 Sep 14. Review.
- Mawrin C, Perry A. Pathological classification and molecular genetics of meningiomas. J Neurooncol. 2010 Sep;99(3):379-91. Epub 2010 Sep 1. Review.
- Gondi V, Tome WA, Mehta MP. Fractionated radiotherapy for intracranial meningiomas. J Neurooncol. 2010 Sep;99(3):349-56. Epub 2010 Sep 1. Review.
- Sheehan JP, Williams BJ, Yen CP. Stereotactic radiosurgery for WHO grade I meningiomas. J Neurooncol. 2010 Sep;99(3):407-16. Epub 2010 Aug 24. Review.
- Sughrue ME, Rutkowski MJ, Aranda D, et al. Treatment decision making based on the published natural history and growth rate of small meningiomas. J Neurosurg. 2010 Nov;113(5):1036-42. Epub 2010 Apr 30. Review.
- Kaye AH, Laws ER. Brain Tumors. New York: Churchill Livingston, 1995.
- Louis DN, Ohgaki H, Wiestler OD, et al. WHO Classification of Tumours of the Central Nervous System. New York: WHO Publications Center, 2007.