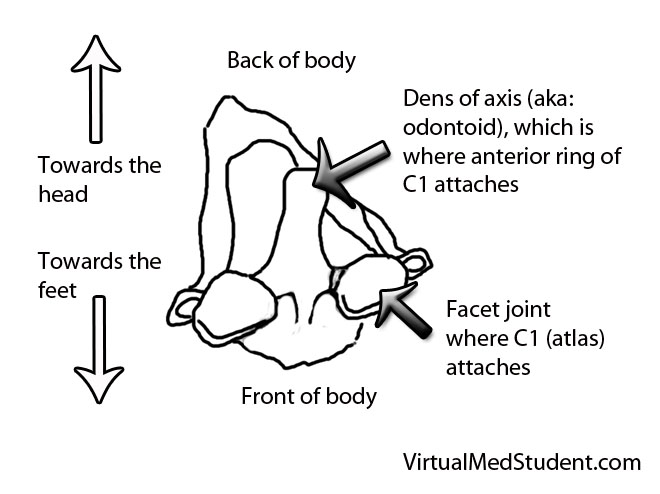
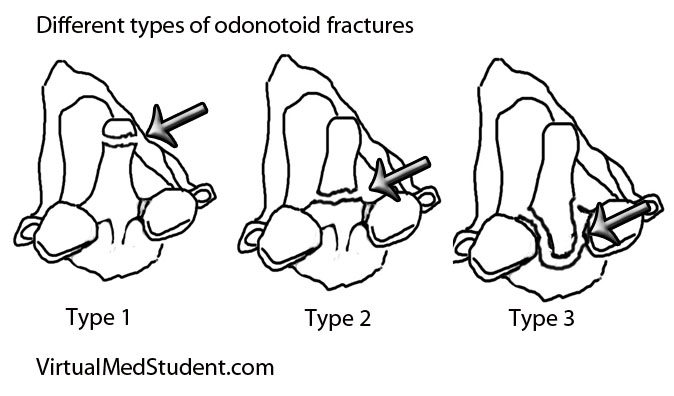
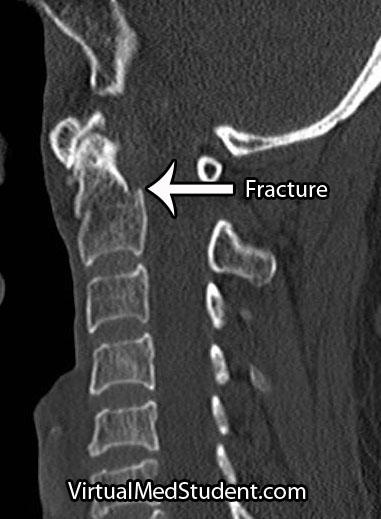
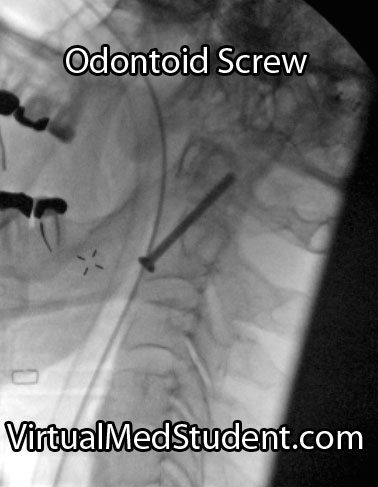
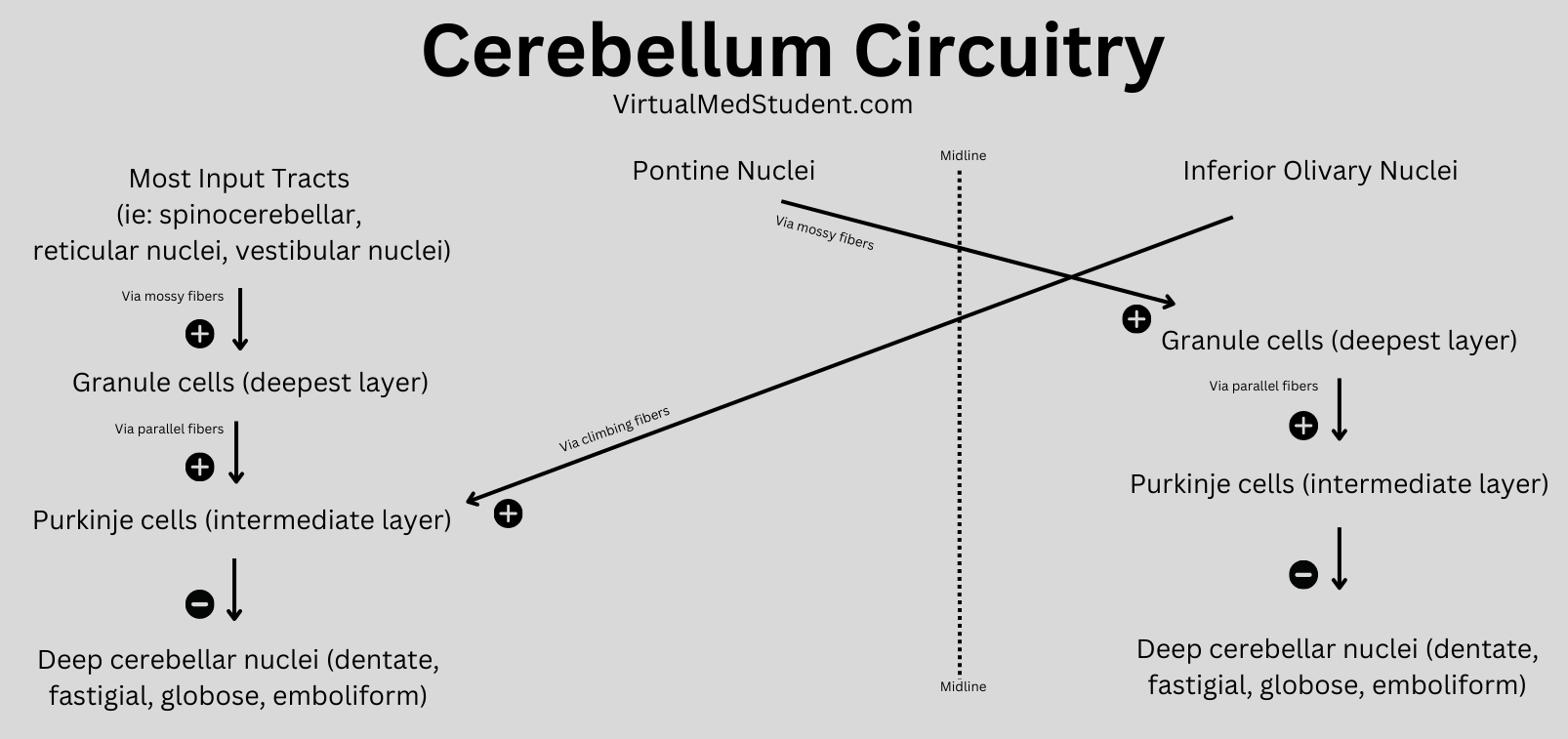
In order to understand what a glioblastoma is we have to first appreciate the different cell types that compose healthy brain tissue. Brain tissue has both neurons and glia. Neurons are the “action” cells of the brain. Glia are the “helper” cells of the brain. They ensure that neurons stay healthy.
A glioblastoma is a malignant brain tumor that arises from a specific type of glial cell known as an astrocyte. Glioblastomas are not only the most common astrocytic tumor, but they are also the most common primary brain tumor!
It is important to realize that there are less malignant tumors that arise from astrocytes (discussed in other articles). Many of these tumors have a much better prognosis, which is why it is important to distinguish glioblastoma from less malignant behaving tumors.
The first distinguishing characteristic is neovascularization. Neovascularization is a fancy medical term used to describe the proliferation of blood vessels within the tumor. As the tumor grows, it requires new vessels to feed it oxygen and nutrients; the process of neovascularization allows the tumor to obtain these essential factors so that it can continue to grow.
Interestingly, as the tumor expands, sections of it will get choked off from its own blood supply. The end result is that part(s) of the tumor actually dies. This is referred to as "necrosis", which is a common finding in glioblastoma.
One of the most distinguishing features of glioblastomas is when cancerous astrocytes "line up" and outline areas of necrosis in a process known as pseudopalisading (see image below).
Given the malignant nature of glioblastoma it is common to see many mitotic figures. Mitotic figures are cells in various states of cell division; these figures indicate a relatively rapidly growing tumor type.
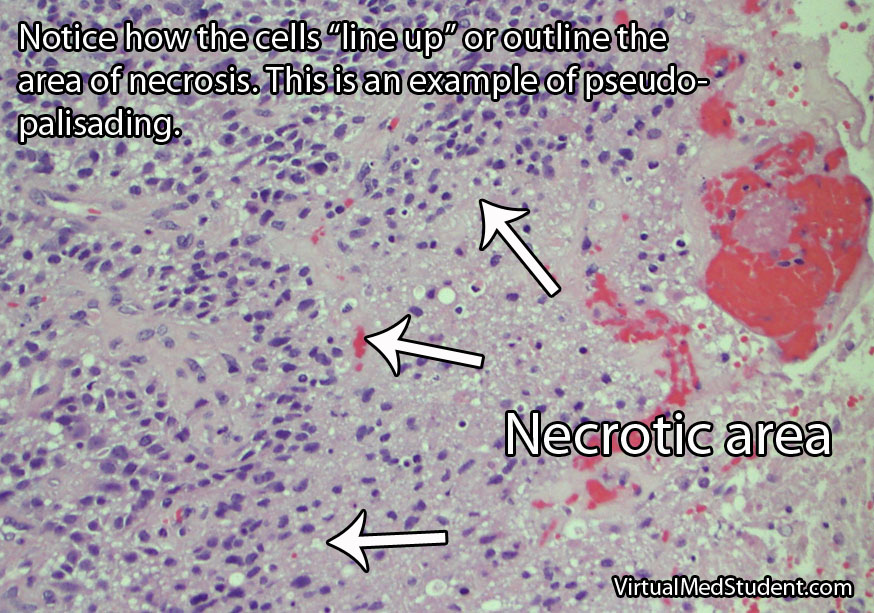
Glibolastoma is therefore characterized by the following pathological characteristics: prominent microvascular proliferation (ie: development of new blood vessels within the tumor), mitosis (ie: an indicator of cell division/growth), and necrosis (ie: areas of dead tumor); pseudopalisading necrosis is a specific form of necrosis shown in the above image that is a hallmark of glioblastomas.
Signs and Symptoms
Glioblastomas may present with any number of signs and/or symptoms depending on their location within the brain. Lesions that are located on the left side of the brain may cause problems with speech if they involve the Broca or Wernicke areas. Tumors in the areas of the brain that control motor movement may cause weakness. Additionally, tumors that arise in the frontal lobes may cause odd behavioral changes. Some patients present with seizures, and others with only a dull headache.
Diagnosis
An official diagnosis of glioblastoma can only be made when a pathologist looks at a sample of the tumor under a microscope. These samples are typically obtained by a neurosurgeon who resects or biopsies the tumor.
However, glioblastomas also have typical features seen on imaging studies such as MRI. For example, these tumors will “rim-enhance” when a contrast material such as gadolinium is infused into the patient during the scan. Rim enhancement is a result of the contrast material leaking out of all of the blood vessels present within the tumor. It is important to note that other diseases such as abscesses, lymphomas, and other infections can also cause rim-enhancement.
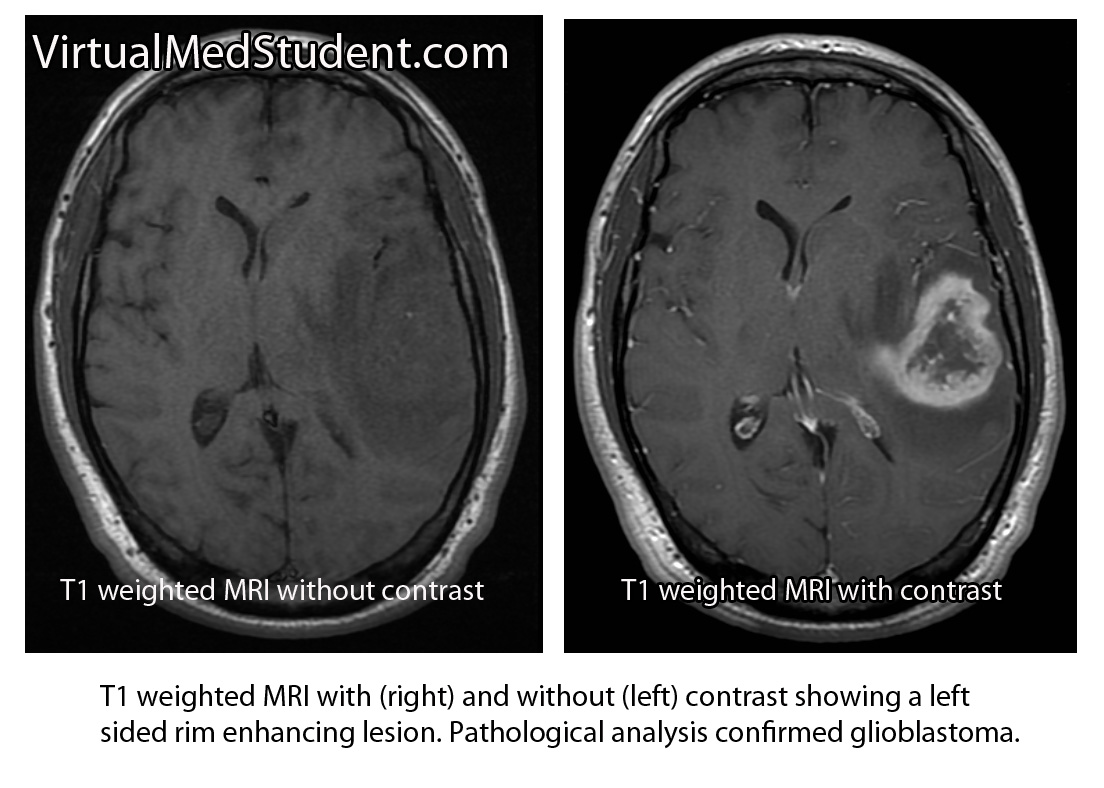
Another useful study known as MR spectroscopy measures the relative amounts of different molecules present within the tumomr. In a glioblastoma the amount of lactate, choline, and lipid are all increased. Lactate is a marker of brain tissue that is not receiving enough oxygen, which is common in necrotic tumor areas. Choline is a molecule that is present in cell membranes. When neurons are rapidly dividing, which is what occurs in glioblastoma, the amount of choline present also increases. A different molecule known as N-acetyl aspartate (NAA) is present in mature cells. Therefore, unlike lactate and choline levels, NAA is decreased in glioblastoma because these cells are "immature" (ie: poorly differentiated).
Treatment
Treatment combines a mixture of surgery, radiation therapy, and different chemotherapeutic drugs, the most common being temozolomide (Temodar®). Surgery is only useful when a significant amount of the tumor can be removed. Despite optimal treatment the prognosis for patients with glioblastoma remains extremely poor.
It is also highly important to treat the edema that frequently surrounds the tumor. Steroids, most commonly dexamethasone (Decadron®), are used to decrease the amount of edema, which usually improves symptoms.
Patients are often started on an anti-seizure medication such as levetiracetam (Keppra®) or phenytoin (Dilantin®).
Overview
Glioblastoma is a malignant astrocytic tumor. It is the most common primary brain tumor. It has unique characteristics that distinguish it from more benign brain tumors that also arise from astrocytes. It is treated with a combination of surgery, radiation, and chemotherapy.
References and Resources
- Wakabayashi T. Clinical trial updates for malignant brain tumors. Rinsho Shinkeigaku. 2011 Nov;51(11):853-6.
- Gulati S, Jakola AS, Nerland US, et al. The risk of getting worse: surgically acquired deficits, perioperative complications, and functional outcomes after primary resection of glioblastoma. World Neurosurg. 2011 Dec;76(6):572-9.
- Lawrence YR, Mishra MV, Werner-Wasik M, et al. Improving prognosis of glioblastoma in the 21st century: Who has benefited most? Cancer. 2011 Dec 16.
- Parlato C, Barbarisi M, Moraci M, et al. Surgery, radiotherapy and temozolomide in treating high-grade gliomas. Front Biosci. 2006 May 1;11:1280-3.
- Wakabayashi T, Kayama T, Nishikawa R, et al. A multicenter phase I trial of combination therapy with interferon-β and temozolomide for high-grade gliomas (INTEGRA study): the final report. J Neurooncol. 2011 Sep;104(2):573-7.