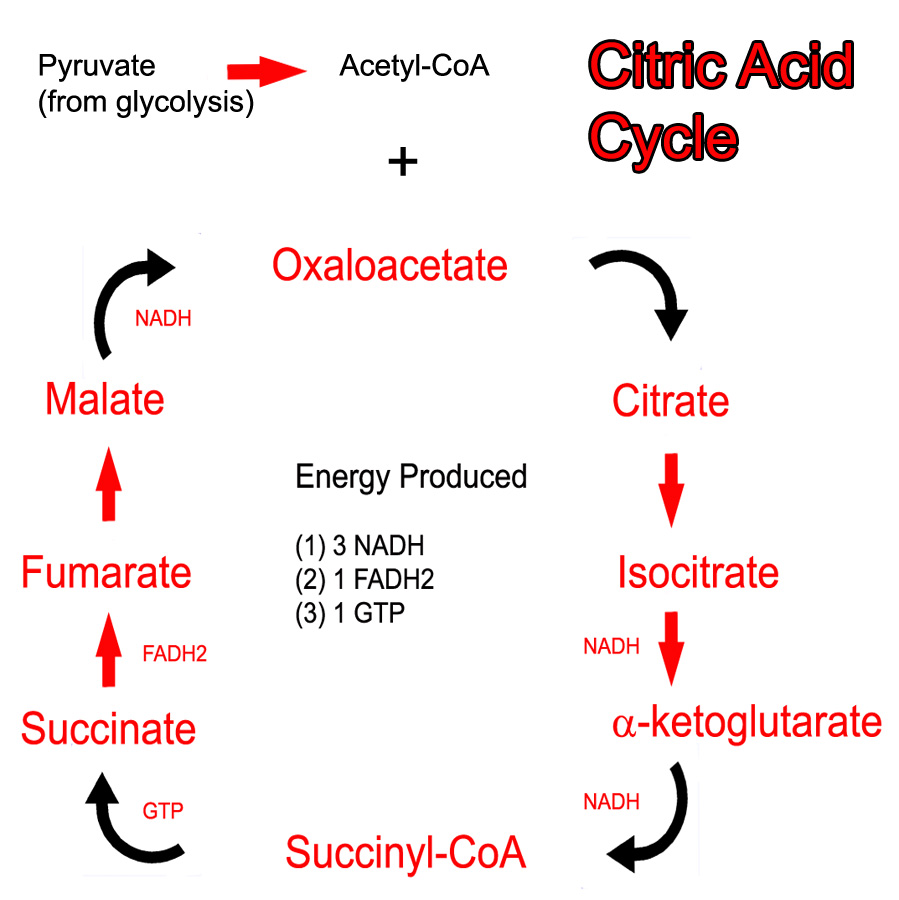
Cellular Anatomy
The cerebellum is a large outgrowth on the backside of the brainstem that looks like a piece of cauliflower. It is the great "modulator" of movement. It compares the movement that the brain wants to do with what the body is actually doing. It then makes fine adjustments to ensure that the intended movement is smooth and fluid. It works in conjunction with the basal ganglia and motor cortex to modulate movement.
As can be expected, the cerebellum is highly complex with inputs and outputs going to various regions of the brain and spinal cord. Before we delve into the circuitry of the cerebellum, we need to first understand its cellular architecture…
Unlike most of the cerebral cortex, the cerebellar cortex has only three layers. They include – from most superficial to deep – the molecular layer, the Purkinje cell layer, and the granular layer.
The molecular layer is composed of connections between the dendrites (ie: information receiving processes) of Purkinje cells and the axons (ie: information sending processes) of granule cells. The molecular layer also contains stellate and basket cells, which help modulate the connections between Purkinje and granule neurons.
The Purkinje layer is, you guessed it, composed of Purkinje neurons. These cells send dendrites into the molecular layer where they receive information from granule cells. Purkinje cells also directly receive signals from other areas of the brain and spinal cord. Purkinje cells then send information along to the deep cerebellar nuclei, which are discrete collections of neurons within the cerebellum.
Finally, the granular layer is populated with granule and Golgi cells. They send axons into the molecular layer, which serve to pass on information to the dendrites of Purkinje cells in the molecular layer.
Confused yet???
Input Circuitry
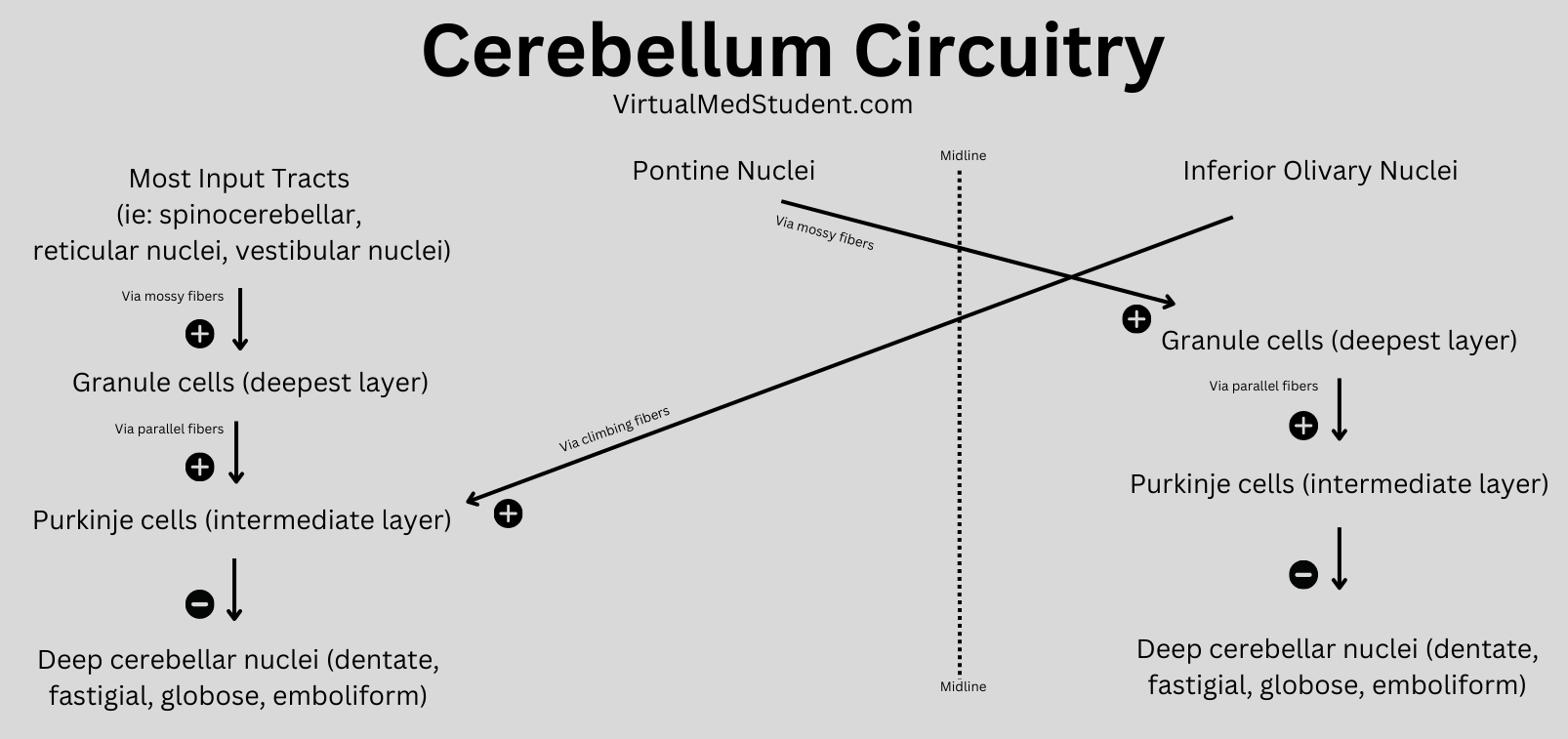
In order for the cerebellum to modulate movement it must receive input from multiple sources. At this point, we have to introduce three new terms: climbing fibers, mossy fibers, and aminergic fibers.
Neurons in an area of the brainstem known as the "olive" (a part of the medulla oblongata) send climbing fibers into the cerebellum. Each climbing fiber forms direct and powerful excitatory connections with multiple Purkinje cells (interestingly, each Purkinje cell only receives input from one climbing fiber).
The second input, mossy fibers, originate from several different areas outside the cerebellum (see table below). These axons form connections with the granule cells. The granule cells, if you remember from our discussion above, send axons into the molecular layer where they form connections to Purkinje cell dendrites. Therefore, mossy fibers indirectly influence Purkinje cells through the "intermediary" granule cells.
Input Connections (ie: Afferent Fibers) to the Cerebellum |
||
| Source of Input | Type of Fiber | Target of Input |
| Olivary nuclei (brainstem) | Climbing fibers | Contralateral cerebellum |
| Pontine nuclei (brainstem) | Mossy fibers | Contralateral cerebellum |
| Reticular nuclei (brainstem) | Mossy fibers | Ipsilateral cerebellum |
| Ventral spinocerebellar tract | Mossy fibers | Ipsilateral cerebellum |
| Dorsal spinocerebellar tract | Mossy fibers | Ipsilateral cerebellum |
| Vestibular nuclei | Mossy fibers | Ipsilateral cerebellum |
| Locus ceruleus | Norepinephrine | Bilateral projections |
| Raphe nucleus | Serotonin | Bilateral projections |
Aminergic fibers originate from the locus ceruleus in the pons, and the raphe nuclei of the midbrain, pons, and medulla. The locus ceruleus fibers "spit" norepinephrine and the raphe nuclei "spit" serotonin onto multiple areas within the cerebellum.
All of these inputs (as well as basket, stellate and Golgi cells, which are intrinsic to the cerebellum itself) are trying to influence the output of the Purkinje neurons. The Purkinje cells ultimately synthesize and pass along all of this competing information via an inhibitory message to the deep cerebellar nuclei.
And that folks brings us to our next section: the output circuitry…
Output from the cerebellum passes exclusively from the deep cerebellar nuclei. The deep nuclei are four discrete collections of neurons, which are given specific (and funky) names; they include the fastigial, globose, emboliform, and dentate nuclei.
Remember that the Purkinje cells inhibit the output of the deep cerebellar nuclei. Therefore, the more active the incoming messages (via mossy and climbing fibers) –> the more active the Purkinje cells –> the less active the output of the deep cerebellar nuclei.
The output of the deep nuclei goes to four major structures outside the cerebellum: the red nucleus, the vestibular nucleus, the reticular formation, and the thalamus. From these structures the information is passed along to the cerebral cortex and/or the spinal cord for additional processing.
Output Connections (ie: Efferent Fibers) from the Cerebellar Nuclei |
||
| Source of Output | Target of Output | Function |
| Globose nucleus | Contralateral red nucleus Contralateral thalamus |
Influences tone of flexor muscles |
| Emboliform nucleus | Contralateral red nucleus Contralateral thalamus |
Influences tone of flexor muscles |
| Dentate nucleus | Contralateral thalamus | Influences motor cortex and coordination |
| Fastigial nucleus | Bilateral vestibular nucleus Bilateral reticular formation |
Influences motor neurons in spinal cord and helps control tone of extensor muscles |
Ultimately, the output of the cerebellum influences not only coordination, but also the tone of flexor and extensor muscles. This allows movement to be smooth and coordinated (unless of course, you are me on the dance floor… in that case all bets are off!).
Overview
The cerebellum is an extremely complex part of the brain. It receives information about an intended movement from the cerebral cortex and compares that to sensory information coming back from the spinal cord. If the intended movement doesn’t match the actual movement the output of the cerebellum attempts to restore balance.
References and Resources
- Ikai Y, Takada M, Shinonaga N, et al. Dopaminergic and non-dopaminergic neurons in the ventral tegmental area of the rat project, respectively, to the cerebellar cortex and deep cerebellar nuclei. Neuroscience, V51:3, p 719-28.
- Asanuma C, Thach WT, Jones EG. Brainstem and spinal projections of the deep cerebellar nuclei in the monkey, with observations on the brainstem projections of the dorsal column nuclei. Brain Research Reviews. V5:3, May 1983. pp 299-322.
- Huang CC, Sugino K, Shima Y, et al. Convergence of pontine and proprioceptive streams onto multimodal cerebellar granule cells. Elife. 2013;2:e00400. Epub 2013 Feb 26.
- Baehr M, Frotscher M. Duus’ Topical Diagnosis in Neurology: Anatomy, Physiology, Signs, Symptoms. Fourth Edition. Stuttgart: Thieme, 2005.
- Purves D, Augustine GJ, Fitzpatrick D, et al. Neuroscience. Fourth Edition. Sinauer Associates, Inc., 2007.