Ogilvie syndrome, which is also known as acute colonic pseudo-obstruction, is a pathological dilation of the colon (ie: “large” intestine) in the absence of a mechanical lesion responsible for the dilation. This differs from colon obstruction caused by tumors, volvulus, strictures, or infection (ie: "mechanical" causes).
The underlying reason why Ogilvie syndrome occurs is not fully understood. The current hypothesis is that the autonomic nervous system, which is partially responsible for gut motility, stops working properly.
Therefore, in order to understand Ogilvie syndrome, we have to understand a little about the autonomic nervous system. The autonomic nervous system is broken up into two parts: sympathetic and parasympathetic. The sympathetic nervous system is responsible for the “fight or flight” response and the parasympathetic system is responsible for the “rest and digest” response. When it comes to the colon, the parasympathetic system increases gut motility and the sympathetic system decreases motility.
Ok, so what is happening in colonic pseudo-obstruction? It is believed that there is an over-abundance of sympathetic input to the colon. This causes a failure of the gut to contract (aka: peristalsis) and therefore feces is not pushed through the GI tract.
If the colon stops contracting then it starts to expand as mucous and stool accumulate. For unknown reasons, the most common part of the colon that is involved in Ogilvie syndrome is the cecum (first portion of the large colon) and the ascending colon.
Signs and Symptoms
Acute colonic pseudo-obstruction causes belly pain, nausea, and vomiting. In addition, since the colon is not contracting, many patients have obstipation (ie: a fancy term for intractable constipation).
If medical attention is not sought soon, the colon may perforate, which can cause peritonitis (infection of the abdominal cavity). Fever, chills, and septic shock may occur in the setting of colonic perforation and peritonitis.
Diagnosis
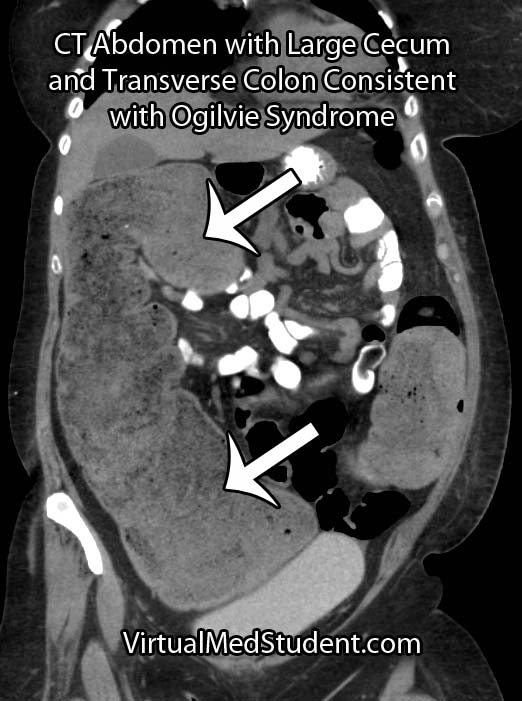
CT scans, abdominal x-rays, barium swallow studies, and colonoscopies are commonly used as adjunctive means for diagnosing pseudo-obstruction.
Treatment
Treatment of Ogilvie syndrome is primarily conservative. This means making the patient NPO (ie: nothing by mouth) and placing a nasogastric tube (NGT) to suction. The NGT helps "decompress" the colon from above until the pseudo-obstruction resolves.
If conservative measures fail then a medication known as neostigmine can be given. Neostigmine is a parasympathetic "mimic". It blocks the breakdown of acetylcholine by inhibiting an enzyme known as acetylcholine esterase. Acetylcholine is the main neurotransmitter of the parasympathetic nervous system. Therefore, if there is more acetylcholine, then more gut motility occurs.
Another medication known as methylnaltrexone may also be useful in cases of acute colonic pseudo-obstruction caused by opioids.
In cases that are refractory to conservative and medical therapies the use of colonoscopic decompression is extremely effective. Patient’s who have evidence of colonic perforation require prompt surgical exploration.
Overview
Ogilvie syndrome, or acute colonic pseudo-obstruction, is a pathologic dilation of the colon that occurs in the absence of a mechanical cause. It is believed to be the result of autonomic nervous system dysfunction. Diagnosis is made with x-rays and CT scans of the abdomen. Treatment is with nasogastric decompression, neostigmine, methylnaltrexone, colonoscopic decompression, or surgery in advanced cases.
References and Resources
- Hsu HL, Wu YM, Liu KL. Ogilvie syndrome: acute pseudo-obstruction of the colon. CMAJ. 2011 February 22; 183(3): E162.
- Eisen GM, Baron TH, Dominitz JA, et al. Acute colonic pseudo-obstruction. Gastrointest Endosc. 2002 Dec;56(6):789-92.
- Saunders MD. Acute colonic pseudo-obstruction. Gastrointest Endosc Clin N Am. 2007 Apr;17(2):341-60, vi-vii.
- Saunders MD, Kimmey MB. Colonic pseudo-obstruction: the dilated colon in the ICU. Semin Gastrointest Dis. 2003 Jan;14(1):20-7.
- Ponec RJ, Saunders MD, Kimmey MB. Neostigmine for the treatment of acute colonic pseudo-obstruction. N Engl J Med. 1999 Jul 15;341(3):137-41.
- Laine L. Management of Acute Colonic Pseudo-Obstruction. N Engl J Med 1999; 341:192-193.
- Thomas J, Karver S, Cooney GA, et al. Methylnaltrexone for opioid-induced constipation in advanced illness. N Engl J Med. 2008 May 29;358(22):2332-43.